Physiopathology of hereditary polyuric states: a molecular view of renal function
DOI: https://doi.org/10.4414/smw.2012.13613
Summary
Recent discoveries have shed new light on the understanding of water metabolism: (1.) in addition to hypothalamic osmoreceptor cells expressing a TRPV1 variant, there are peripheral TRPV4 receptors sensing tonicity in the portal vein and changing central vasopressin secretion and peripheral autonomic activity; (2.) the central osmoregulatory gain of angiotensin action participates in the non-osmotic release of vasopressin induced by hypovolaemia; (3.) prostaglandins EP2 receptors on principal cells of the collecting ducts positively regulate urine concentration mechanisms. These new developments are important clinically for the understanding of hereditary polyuric states. We recommend sequencing of the nephrogenic diabetes insipidus genes in all affected patients. This genomic information is key to the routine care of patients with congenital polyuria and, as in other genetic diseases, reduces health costs and confers psychological benefits on patients and families.
Introduction
Over and above the multifactorial processes of excretion, brain circumventricular sensors (fig. 1) reacting to tonicity subserve the control of antidiuretic hormone secretion and thirst. Hypertonicity can mediate a proportional excitation of the brain’s primary osmoreceptor neurons via mechanical modulation of nonselective cation channels encoded by Trpv1 (transient receptor potential vanilloid-1 gene) [1]. Specifically, hypertonicity is sensed by organum vasculosum lamina terminalis (OVLT) neurons expressing TRPV1: OVLT serves as the brain's primary osmoreceptor area [2] and neurons in this nucleus transduce hyperosmotic conditions into proportional increases in action potential firing rate [3]. The information encoded by the electrical activity of these neurons is then relayed synaptically to diverse subsets of homeostatic effector neurons that induce appropriate osmoregulatory responses such as thirst, natriuresis and antidiuretic hormone release [4–8].
In this article I describe central (OVLT) and peripheral (hepatic neurons) osmoreceptors respectively expressing TRPV1 and TRPV4 cation channels, both critical in maintaining plasma osmolality within normal limits through control of antidiuretic hormone secretion and thirst.
I also explain the effect of vasopressin on principal cells of the renal collecting ducts in generating intracellular cyclic AMP and the insertion of the AQP2 water channels into the luminal membrane, a necessary step for distal water reabsorption.
I use these new physiopathological data to explain hereditary central and nephrogenic diabetes insipidus and to facilitate their early clinical recognition and molecular evaluation.
In a recent editorial Richard Lifton indicated that mutations causing more than 2000 Mendelian diseases have been identified, and this has led to rewriting of of pathophysiology textbooks on every organ system and the identification of rational targets for therapeutic intervention [9]. I use the new information provided by the AVP, AVPR2 and AQP2 genes to describe a molecular view of water conservation.
Plasma sodium and osmolality
Plasma sodium and osmolality are maintained within normal limits (136–143 mEq/l for plasma sodium; 275–290 mOsmol/kg for plasma osmolality) by a thirst-AVP-renal axis [7, 10]. Thirst and AVP release, both stimulated by increased osmolality, is a “double negative” feedback system [11]. Even when the AVP component of this “double negative” regulatory feedback system is lost, the thirst mechanism still preserves plasma sodium and osmolality within the normal range, but at the expense of pronounced polydipsia and polyuria. Thus, the plasma sodium concentration or osmolality of an untreated patient with diabetes insipidus and unlimited access to water may be slightly greater than the mean normal value, and a decrease in plasma sodium and osmolality might be observed in primary polydipsic patients, though these small increases have no diagnostic significance [12].
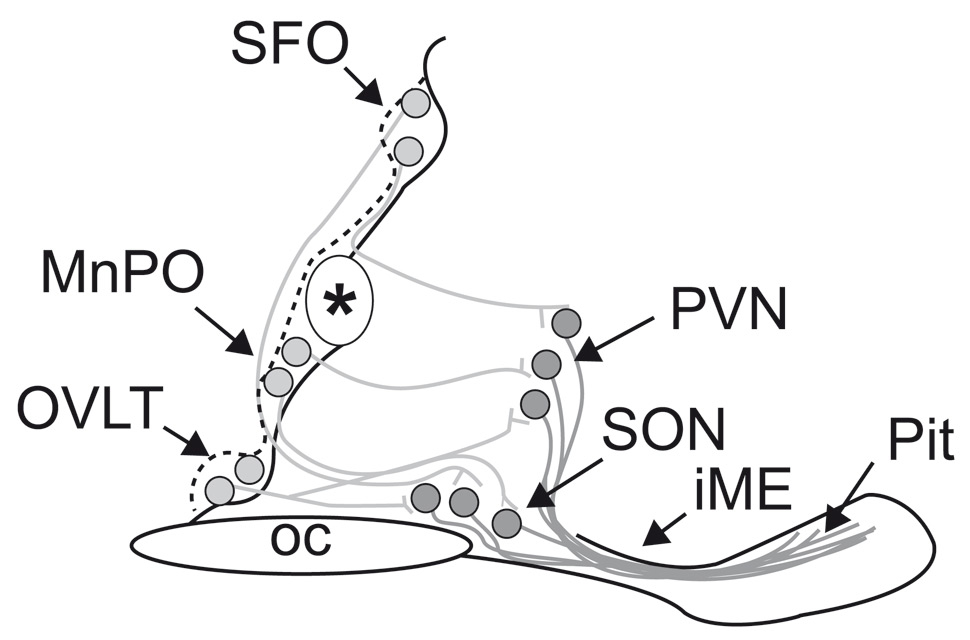
Figure 1
Schematic representation of the osmoregulatory pathway of the hypothalamus (sagittal section of midline of ventral brain around the 3rd ventricle in mice). Neurons (lightly filled circles) in the lamina terminalis (OVLT), median preoptic nucleus (MnPO) and subfornical organ (SFO) – that are responsive to plasma hyptertonicity send efferent axonal projections (grey lines) to magnocellular neurons of the paraventricular (PVN) and supraoptic nuclei (SON). The OVLT is one of the brain circumventricular organs and is a key osmosensing site in the mammalian brain (vide infra). The processes (dark lines) of these magnocellular neurons form the hypothalamo-neurohypophysial pathway that courses in the median eminence to reach the posterior pituitary, where neurosecretion of vasopressin and oxytocin occurs. (Modified from [80]).
Mammals are osmoregulators: the cellular perception of tonicity
Mammals are osmoregulators: they have evolved mechanisms that maintain extracellular fluid (ECF) osmolality near a stable value. But, although mammals strive to maintain a constant ECF osmolality, values measured in an individual can fluctuate around the set-point owing to intermittent changes in the rates of water intake and water loss (through evaporation or diuresis) and to variations in the rates of Na intake and excretion (natriuresis). In humans, for example, 40 minutes of strenuous exercise in the heat [13, 14], or 24 hours of water deprivation, [15] causes plasma osmolality to rise by more than 10 mosmol/kg. In a dehydrated individual, drinking the equivalent of two large glasses of water (~850 ml) lowers osmolality by approximately 6 mosmol/kg within 30 minutes [16]. Similarly, ingestion of 13 g salt increases plasma osmolality by approximately 5 mosmol/kg within 30 minutes [17]. Although osmotic perturbations larger than these can be deleterious to health, changes in the 1–3% range play an integral part in the control of body-fluid homeostasis. Differences between the ECF osmolality and the desired set-point induce proportional homeostatic responses according to the principle of negative feedback [7]. ECF hyperosmolality stimulates the sensation of thirst, to promote water intake, and the release of vasopressin to enhance water reabsorption in the kidney. By contrast, ECF hypoosmolality suppresses basal VP secretion in rats and humans [18].
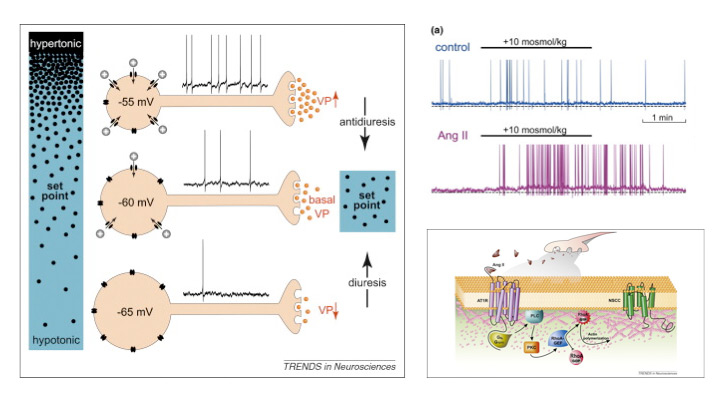
Figure 2
Upper left: Cell autonomous osmoreception in vasopressin neurons. Changes in osmolality cause inversely proportional changes in soma volume. Shrinkage activates nonselective cation channels (NSCCs) and the ensuing depolarisation increases action potential firing rate and vasopressin (VP) release from axon terminals in the neurohypophysis. Increased VP levels in blood enhance water reabsorption by the kidney (antidiuresis) to restore extracellular fluid osmolality toward the set point. Hypotonic stimuli inhibit NSCCs. The resulting hyperpolarization and inhibition of firing reduces VP release and promotes diuresis.
Upper right: Whole cell current clamp recordings from isolated MNCs (left) and averaged data from multiple cells show that the depolarising and action potential firing responses induced by a hypertonic stimulus are significantly enhanced in the presence of 100 nM Ang II.
Lower right: Hypothetical events mediating central Ang II enhancement of osmosensory gain. Ang II released by afferent nerve terminals (e.g. during hypovolaemia) binds to AT1 receptor (AT1R) coupled to G proteins such as Gq or/and G12/13. Activated G proteins signal through phospholipase C (PLC) and protein kinase C (PKC) to activate a RhoA-specific guanine nucleotide exchange factor (RhoA–GEF), such as p115RhoGEF or LARG (leukaemia-associated Rho guanine–nucleotide exchange factor) Activation of RhoA–GEF converts inactive cytosolic RhoA (RhoA–GDP) into active, membrane-associated RhoA–GTP by promoting the exchange of GDP to GTP. Activated RhoA induces actin polymerisation and increases submembrane F-actin density to enhance the mechanical gating of non-specific cation channels.
(From [81]: Prager-Khoutorsky M, Bourque CW. Osmosensation in vasopressin neurons: changing actin density to optimize function. Trends Neurosci. 2010;33:76–83. © Elsevier, Oxford, UK, with kind permission.)
As summarised elegantly by Bourque [7], early studies provided clear evidence that “cellular dehydration” (that is, cell shrinking) was required to stimulate thirst and vasopressin release during ECF hyperosmolality: these responses could be induced by infusions of concentrated solutions containing membrane-impermeable solutes, which extract water from cells, but not by infusions of solutes that readily equilibrate across the cell membrane (such as urea). Verney coined the term osmoreceptor to designate the specialised sensory elements. He further showed that these were present in the brain and postulated that they might comprise “tiny osmometers” and “stretch receptors” that would allow osmotic stimuli to be “transmuted into electrical signals” [19]. Osmoreceptors are therefore defined functionally as neurons that are endowed with an intrinsic ability to detect changes in ECF osmolality and it is now known that both cerebral and peripheral osmoreceptors contribute to the body fluid balance (fig. 2).
Although magnocellular neurons are themselves osmosensitive, they require input, by glutamatergic afferents, from the lamina terminalis in order to respond fully to osmotic challenges (fig. 1). Neurons in the lamina terminalis are also osmosensitive and because the subfornical organ (SFO) and the organum vasculosum of the lamina terminalis (OVLT) lie outside the blood-brain barrier, they can integrate this information with endocrine signals borne by circulating hormones, such as angiotensin II (Ang-II), relaxin, and atrial natriuretic peptide (ANP). While circulating Ang-II and relaxin excite both OT and vasopressin magnocellular neurons, ANP inhibits vasopressin neurons. The non-osmotic pathways are more physiologically described now as “osmoregulatory gain” since angiotensin II amplifies osmosensory transduction by enhancing the proportional relationship between osmolality, receptor potential and action potential firing in rat supraoptic nucleus neurons [20]. Modifications in osmoregulatory gain induced by angiotensin explain why the changes in the slope and threshold of the relationship between plasma osmolality and vasopressin secretion are potentiated by hypovolaemia or hypotension and are attenuated by hypervolaemia or hypertension [21] (fig. 3).
Tonicity information is relayed by central osmoreceptor neurons expressing TRPV1 and peripheral osmoreceptor neurons expressing TRPV4
Osmotic regulation of the release of AVP from the posterior pituitary is primarily dependent, under normal circumstances, on tonicity information relayed by central osmoreceptor neurons expressing TRPV1 [7] and peripheral osmoreceptor neurons expressing TRPV4 [10].
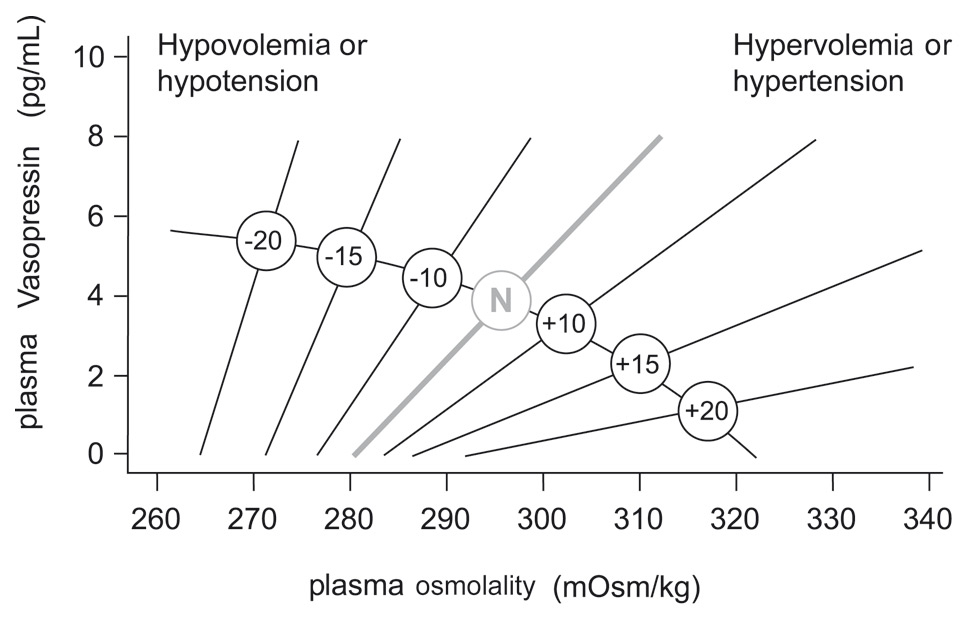
Figure 3
Schematic representation of the relationship between plasma vasopressin and plasma osmolality in the presence of differing states of blood volume and/or pressure.
The line labelled N represents normovolaemic normotensive conditions. Minus numbers to the left indicate percent fall, and positive numbers to the right, percent rise in blood volume or pressure.
The cellular basis for osmoreceptor potentials has been characterised using patch-clamp recordings and morphometric analysis in magnocellular cells isolated from the supraoptic nucleus of the adult rat. In these cells, stretch-inactivating cationic channels transduce osmotically evoked changes in cell volume into functionally relevant changes in membrane potential. In addition, magnocellular neurons also operate as intrinsic Na+ detectors. The N-terminal variant of the transient receptor potential channel (TRPV1) is an osmotically activated channel expressed in the magnocellular cells producing vasopressin [22] and in the circumventricular organs, the OVLT and the SFO [3]. Since osmoregulation still operates in Trpv1−/− mice, other osmosensitive neurons or pathways must be able to compensate for loss of central osmoreceptor function [3, 22, 23]. Afferent neurons expressing the osmotically activated ion channel, TRPV4 in the thoracic dorsal root ganglia that innervate hepatic blood vessels and detect physiological hypo-osmotic shifts in blood osmolality have recently been identified [10]. In mice lacking the osmotically activated ion channel, TRPV4, hepatic sensory neurons no longer exhibit osmosensitive inward currents and activation of peripheral osmoreceptors in vivo is abolished. In a large cohort of human liver transplantees, who presumably have denervated livers, plasma osmolality is significantly elevated compared to healthy controls, suggesting the presence of an inhibitory vasopressin effect of hyponatraemia, perceived in the portal vein from hepatic afferents [10]. TRPV1 (expressed in central neurons) and TRPV4 (expressed in peripheral neurons) thus appear to play entirely complementary roles in osmoreception. Lechner et al., have thus identified the primary afferent neurons that constitute the afferent arc of a well-characterised reflex in man and more recently also in rodents [24]. This reflex engages the sympathetic nervous system to raise blood pressure and stimulate metabolism [25, 26]. As a point of clinical interest, it has already been demonstrated that orthostatic hypotension and postprandial hypotension respond to water drinking [27–29]. Moreover, water drinking in man can prevent neutrally mediated syncope during blood donation or after prolonged standing [30]. Finally, water drinking is also associated with weight loss in overweight individuals [31]. Other peripheral sensory neurons expressing other mechanosensitive proteins may also be involved in osmosensitivity [32].
Arginine vasopressin synthesis
AVP and its corresponding carrier, neurophysin II, are synthesised as a composite precursor by the magnocellular neurons of the supraoptic and paraventricular nuclei of the hypothalamus (for review see [33] fig. 4). The precursor is packaged into neurosecretory granules and transported axonally in the stalk of the posterior pituitary. En route to the neurohypophysis, the precursor is processed into the active hormone. Prepro-vasopressin has 164 amino acids and is encoded by the 2.5 kb AVP gene located in chromosome region 20p13 [34, 35]. The AVP gene (coding for AVP and neurophysin II) and the OXT gene (coding for oxytocin and neurophysin I) are located in the same chromosome region, at a very short distance from each other (12 kb in humans) in head to head orientation. Data from transgenic mouse studies indicate that the intergenic region between the OXT and the AVP genes contains the critical enhancer sites for cell-specific expression in the magnocellular neurons [33]. It is phylogenetically interesting to note that cis and trans components of this specific cellular expression have been conserved between the Fugu isotocin (the homologue of mammalian oxytocin) and rat oxytocin genes [36]. Exon 1 of the AVP gene encodes the signal peptide, AVP, and the NH2-terminal region of neurophysin II. Exon 2 encodes the central region of neurophysin II, and exon 3 encodes the COOH-terminal region of neurophysin II and the glycopeptide. Pro-vasopressin is generated by removal of the signal peptide from prepro-vasopressin and from the addition of a carbohydrate chain to the glycopeptide (fig. 4). Additional post-translation processing occurs within neurosecretory vesicles during transport of the precursor protein to axon terminals in the posterior pituitary, yielding AVP, neurophysin II, and the glycopeptide. The AVP-neurophysin II complex forms tetramers that can self-associate to form higher oligomers [37]. Neurophysins should be seen as chaperone-like molecules facilitating intracellular transport in magnocellular cells. In the posterior pituitary, AVP is stored in vesicles. Exocytotic release is stimulated by minute increases in serum osmolality (hypernatraemia, osmotic regulation) and by more pronounced decreases in extracellular fluid (hypovolaemia, non-osmotic regulation). Oxytocin and neurophysin I are released from the posterior pituitary by the suckling response in lactating females.
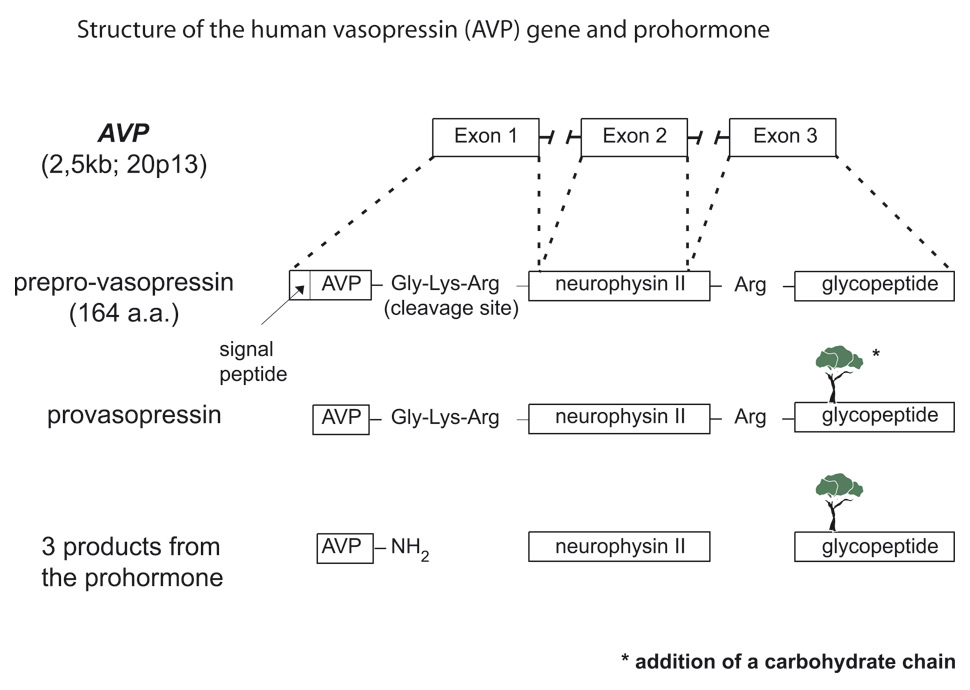
Figure 4
Structure of the human vasopressin (AVP) gene and cascade of vasopressin biosynthesis, signal peptide; AVP, arginine-vasopressin; neurophysin; glycoprotein.
Immunocytochemical and radioimmunological studies have demonstrated that oxytocin and vasopressin are synthesised in separate populations of the supraoptic nuclei and the paraventricular nuclei neurons [38, 39], whose central and vascular projections have been described in great detail [40]. Some cells express the AVP gene and others express the OXT gene. Immunohistochemical studies have revealed a second vasopressin neurosecretory pathway that transports high concentrations of the hormone to the anterior pituitary gland from parvocellular neurons to the hypophyseal portal system. In the portal system, the high concentration of AVP acts synergistically with corticotropin-releasing hormone (CRH) to stimulate adrenocorticotropin (ACTH) release from the anterior pituitary. More than half of parvocellular neurons coexpress both CRH and AVP. In addition, while passing through the median eminence and the hypophyseal stalk, magnocellular axons can also release AVP into the long portal system. Furthermore, a number of neuroanatomical studies have revealed the existence of short portal vessels that allow communication between the posterior and anterior pituitary. Thus, in addition to parvocellular vasopressin, magnocellular vasopressin is able to influence ACTH secretion [41, 42].
Cellular actions of vasopressin
The neurohypophyseal hormone AVP has multiple actions, including inhibition of diuresis, contraction of smooth muscle, platelet aggregation, stimulation of liver glycogenolysis, modulation of ACTH release from the pituitary, and central regulation of somatic functions (thermoregulation, blood pressure) [43]. These multiple actions of AVP can be explained by the interaction of AVP with at least three types of G protein-coupled receptors; the V1a (vascular hepatic) and V1b (anterior pituitary) receptors act through phosphatidylinositol hydrolysis to mobilise calcium [44], and the V2 (kidney) receptor is coupled to adenylate cyclase [45]. Oxytocin and vasopressin receptors are widely expressed in the brain and it is now well recognised that oxytocin and vasopressin are involved in adult-adult attachment [46] and in the modulation of autonomic fear responses [47].
The first step in the action of AVP on water excretion is its binding to arginine vasopressin type 2 receptors (hereafter referred to as V2 receptors) on the basolateral membrane of the collecting duct cells (fig. 5). The human gene that codes for the V2 receptor (AVPR2) is located in chromosome region Xq28 and has three exons and two small introns [48, 49]. The sequence of the cDNA predicts a polypeptide of 371 amino acids with seven transmembrane, four extracellular, and four cytoplasmic domains. The V2 receptor is one of 701 members of the rhodopsin family within the superfamily of guanine-nucleotide (G) protein-coupled receptors [50]; see also perspective by [51]. The activation of the V2 receptor on renal collecting tubules stimulates adenylyl cyclase via the stimulatory G protein (Gs) and promotes the cyclic adenosine monophosphate (cAMP)-mediated incorporation of water pores into the luminal surface of these cells. This process is the molecular basis of the vasopressin-induced increase in the osmotic water permeability of the apical membrane’s collecting tubule [52–54].
AVP also increases the water reabsorptive capacity of the kidney by regulating the urea transporter UT-A1 that is present in the inner medullary collecting duct, predominantly in its terminal part [55, 56]. AVP also increases the permeability of principal collecting duct cells to sodium [57].
In summary, in the absence of AVP stimulation, collecting duct epithelia exhibit very low permeabilities to sodium urea and water. These specialised permeability properties permit the excretion of large volumes of hypotonic urine formed during intervals of water diuresis. By contrast, AVP stimulation of the collecting ducts’ principal cells leads to selective increases in the permeability of the apical membrane to water, urea and sodium.
These actions of vasopressin in the distal nephron are possibly modulated by prostaglandin E2, nitric oxide [58], and by luminal calcium concentration. PGE2 is synthesised and released in the collecting duct, which expresses all four E-prostanoid receptors (EP1–4). Both EP2 and EP4 can signal via increased cAMP. Olesen et al. hypothesised that selective EP receptor stimulation could mimic the effects of vasopressin and demonstrated that, at physiological levels, PGE2 markedly increased apical membrane abundance and phosphorylation of AQP2 in vitro and ex vivo, leading to increased cell water permeability [59]. In their experiments, both EP2 and EP4 selective agonists were able to mimic these effects. Furthermore, an EP2 agonist was able to regulate positively urinary-concentrating mechanisms in an animal model of nephrogenic diabetes insipidus (NDI). These results reveal an alternative mechanism for regulating water transport in the collecting duct that has major importance for understanding whole-body water homeostasis and provides a rationale for investigations into EP receptor agonist use in nephrogenic diabetes insipidus treatment. An apical calcium/polycation receptor protein expressed in the terminal portion of the rat inner medullary collecting duct has been shown to reduce AVP-elicited osmotic water permeability when luminal calcium concentration rises [60]. This possible link between calcium and water metabolism may play a role in the pathogenesis of renal stone formation [60].
Hereditary central and nephrogenic diabetes insipidus (table 1)
Diabetes insipidus is a disorder characterised by the excretion of abnormally large volumes (greater than 30 ml/kg body weight per day for an adult patient) of dilute urine (less than 250 mmol/kg). This definition excludes osmotic diuresis, which occurs when excess solute is being excreted, e.g. glucose in the polyuria of diabetes mellitus. Other agents that produce osmotic diuresis are mannitol, urea, glycerol, contrast media, and loop diuretics. Osmotic diuresis should be considered when solute excretion exceeds 60 mmol/hour. Four basic defects may be involved. The most common, deficient secretion of the antidiuretic hormone (ADH) arginine vasopressin (AVP), is referred to as neurogenic (or central, neurohypophyseal, cranial, or hypothalamic) diabetes insipidus. Diabetes insipidus may also result from renal insensitivity to the antidiuretic effect of AVP, which is referred to as nephrogenic diabetes insipidus. Excessive water intake may result in polyuria, which is referred to as primary polydipsia; it may be due to an abnormality in the thirst mechanism, referred to as dipsogenic diabetes insipidus, or it may be associated with a severe emotional cognitive dysfunction, referred to as psychogenic polydipsia. Finally, increased metabolism of vasopressin during pregnancy is referred to as gestational diabetes insipidus.
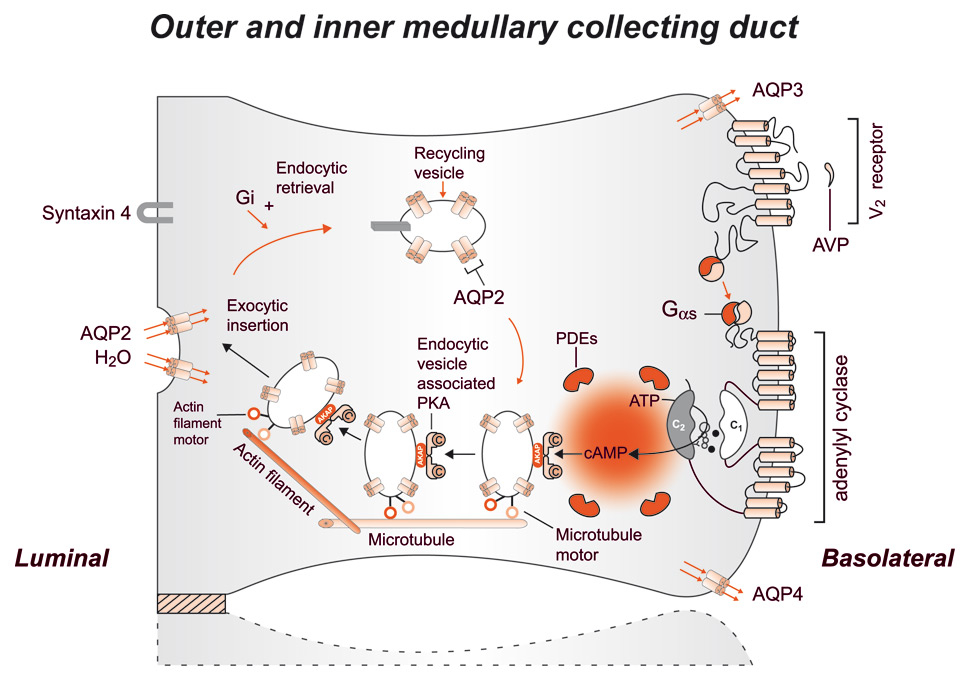
Figure 5
Schematic representation of the effect of vasopressin (AVP) to increase water permeability in the principal cells of the collecting duct. AVP is bound to the V2 receptor (a G-protein-linked receptor) on the basolateral membrane. The basic process of G-protein-coupled receptor signalling consists of three steps: a hepta-helical receptor that detects a ligand (in this case, AVP) in the extracellular milieu, a G-protein (Gαs) that dissociates into α subunits bound to GTP and βγ subunits after interaction with the ligand-bound receptor, and an effector (in this case, adenylyl cyclase) that interacts with dissociated G-protein subunits to generate small-molecule second messengers. AVP activates adenylyl cyclase, increasing the intracellular concentration of cAMP. The topology of adenylyl cyclase is characterised by two tandem repeats of six hydrophobic transmembrane domains separated by a large cytoplasmic loop and terminates in a large intracellular tail. The dimeric structure (C1 and C2) of the catalytic domains is represented. Conversion of ATP to cAMP takes place at the dimer interface. Two aspartate residues (in C1) coordinate two metal co-factors (Mg2+ or Mn2+ represented here as two small black circles), which enable the catalytic function of the enzyme (305). Adenosine is shown as an open circle and the three phosphate groups (ATP) are shown as smaller open circles. Protein kinase A (PKA) is the target of the generated cAMP. The binding of cAMP to the regulatory subunits of PKA induces a conformational change, causing these subunits to dissociate from the catalytic subunits. These activated subunits (C) as shown here are anchored to an aquaporin-2 (AQP2)-containing endocytic vesicle via an A-kinase anchoring protein. The local concentration and distribution of the cAMP gradient is limited by phosphodiesterases (PDEs). Cytoplasmic vesicles carrying the water channels (represented as homotetrameric complexes) are fused to the luminal membrane in response to AVP, thereby increasing the water permeability of this membrane. The dissociation of the A-kinase anchoring protein from the endocytic vesicle is not represented. Microtubules and actin filaments are necessary for vesicle movement toward the membrane. When AVP is not available, AQP2 water channels are retrieved by an endocytic process, and water permeability returns to its original low rate. Aquaporin-3 (AQP3) and aquaporin-4 (AQP4) water channels are expressed constitutively at the basolateral membrane.
(From: Bonnardeaux A, Bichet DG. Inherited disorders of the renal tubule. In: Brenner BM (ed). The Kidney. 8th ed. New York: Harcourt Health Sciences; 2008. p. 1382-1419. © Elsevier, Oxford, UK, with kind permission.)
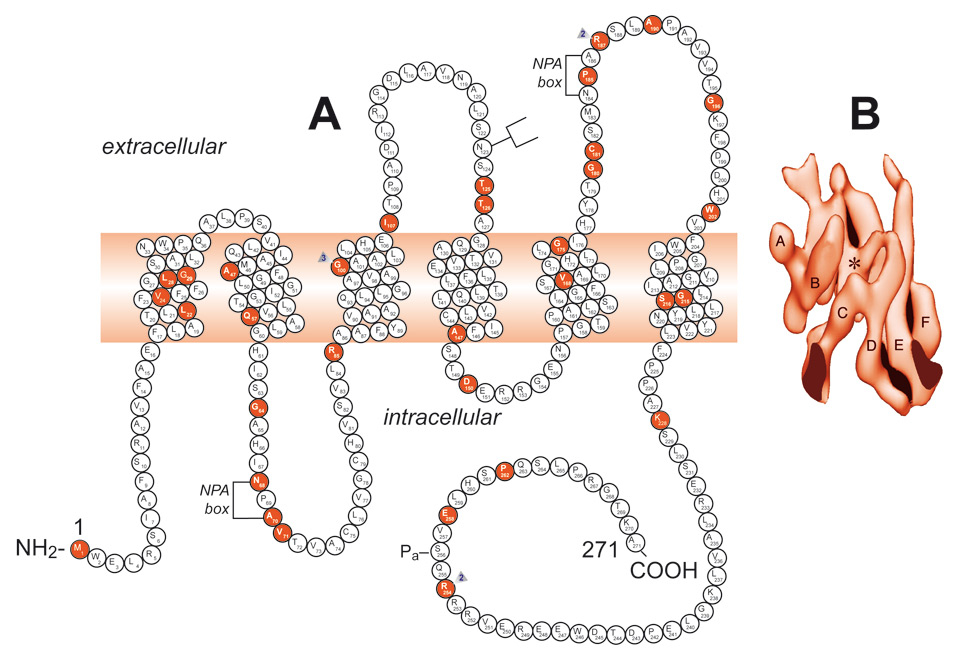
Figure 6
A Schematic representation of the aquaporin-2 protein and identification of 46 AQP2 mutations. A monomer is represented with six transmembrane helices. The location of the protein kinase A phosphorylation site (Pa) is indicated. The extracellular, transmembrane, and cytoplasmic domains are defined according to Deen et al. [82] Solid symbols indicate the mutations’ location.
B A surface-shaded representation of the six-helix barrel of the AQP1 protein viewed parallel to the bilayer.
(From: Bonnardeaux A, Bichet DG. Inherited disorders of the renal tubule. In: Brenner BM (ed). The Kidney. 8th ed. New York: Harcourt Health Sciences; 2008. p. 1382‒1419. © Elsevier, Oxford, UK, with kind permission.)
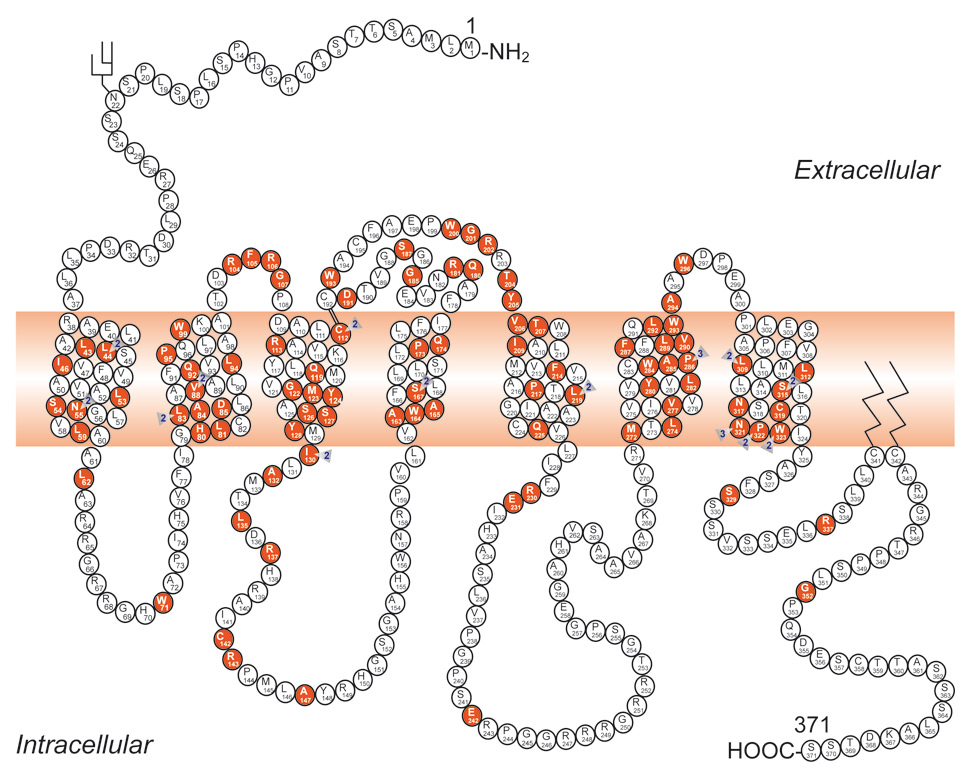
Figure 7
Schematic representation of the V2 receptor and identification of 193 putative disease-causing AVPR2 mutations. Predicted amino acids are shown as the one-letter amino acid code. A solid symbol indicates a codon with a missense or nonsense mutation; a number indicates more than one mutation in the same codon; other types of mutation are not indicated on the figure. There are 95 missense, 18 nonsense, 46 frameshift deletion or insertion, 7 inframe deletion or insertion, 4 splice-site, and 22 large deletion mutations, and one complex mutation.
(From: Bonnardeaux A, Bichet DG. Inherited disorders of the renal tubule. In: Brenner BM (ed). The Kidney. 8th ed. New York: Harcourt Health Sciences; 2008. p. 1382‒1419. © Elsevier, Oxford, UK, with kind permission.)

Figure 8
A young patient thirsty since birth.
Hereditary central diabetes insipidus
Most of the central diabetes insipidus cases seen in general practice are acquired, but the rare cases of hereditary autosomal dominant or recessive neurohypophyseal diabetes insipidus have provided further cellular understanding of the mechanisms responsible for pre-hormone folding, maturation and release. Autosomal dominant central diabetes insipidus is secondary to the toxic accumulation of vasopressin mutants as fibrillar aggregates in the endoplasmic reticulum of hypothalamic magnocellular neurons producing vasopressin.
Inherited neurohypophyseal diabetes insipidus (OMIM 125700) [61] due to mutations in the AVP gene (OMIM 192340) [61]
Patients with autosomal dominant neurohypophyseal diabetes insipidus retain some limited capacity to secrete AVP during severe dehydration, and the polyuria-polydipsic symptoms usually appear after the first year of life [62], when the infant’s demand for water is more likely to be understood by adults. In neurohypophyseal diabetes insipidus, termed familial neurohypophyseal diabetes insipidus (FNDI), levels of AVP are insufficient and patients show a positive response to treatment with dAVP. Growth retardation may be observed in untreated children with autosomal dominant FNDI [63]. Over 60 mutations in the prepro-arginine-vasopressin-neurophysin II AVP gene located on chromosome 20p13 have been reported in dominant FNDI (adFNDI). Knock-in mice heterozygous for a nonsense mutation in the AVP carrier protein neurophysin II showed progressive loss of AVP-producing neurons over several months correlated with increased water intake, increased urine output, and decreased urine osmolality. The data suggest that vasopressin mutants accumulate as fibrillar aggregates in the endoplasmic reticulum and cause cumulative toxicity to magnocellular neurons explaining the later age of onset [64, 65]. To date, recessive FNDI, with early polyuric manifestations has only been described in three studies [66–68]. Very early (first week of life) polyuric states are usually nephrogenic but we and others have observed autosomal recessive central diabetes insipidus patients with early polyuria, dehydration episodes responding to dDAVP with specific mutations of the AVP gene [66–69]. A study by Christensen et al. [70] examined the differences in cellular trafficking between dominant and recessive AVP mutants and found that dominant forms were concentrated in the cytoplasm, whereas recessive forms were localised to the tips of neurites. The expression of regulated secretory proteins such as granins and prohormones, including pro-vasopressin, generates granule-like structures in a variety of neuroendocrine cell lines due to aggregation in the trans-Golgi [71]. Co-staining experiments unambiguously distinguished between these granule-like structures and the accumulations by pathogenic dominant mutants formed in the ER, since the latter, but not the trans-Golgi granules, co-localised with specific ER markers [64]. As studies concerning both dominant and recessive FNDI accumulate, it is becoming evident that FNDI exhibits a variable age of onset and this may be related to the cellular handling of the mutant AVP. This progressive toxicity, sometimes called a toxic gain of function, shares mechanistic pathways with other neurodegenerative diseases such as Huntington’s and Parkinson’s.
It is of interest that no loss of function of the TRPV1 or TRPV4 genes has been linked to a defect in water excretion in humans, yet a rare polymorphism of TRPV4 has been associated with human hyponatraemia [72].
|
Table 1
|
|
Hereditary central and nephrogenic diabetes insipidus
|
| ↓
Hereditary central diabetes insipidus
|
↓
Hereditary nephrogenic diabetes insipidus
|
|
Clinical features
|
Polyuria, polydipsia, low urine osmolality during dehydration, increased urine osmolality after desamino-8D-arginine vasopressin (dDAVP) |
Polyuria, polydipsia, low urine osmolality during dehydration, but little or no increase in urine osmolality after dDAVP |
|
Genetic analysis
|
Mutations in the AVP gene, most are autosomal dominant |
↓
X-linked:
AVPR2 mutations, males affected but also expressing females |
↓
Autosomal dominant or recessive: AQP2 mutations |
Hereditary nephrogenic diabetes insipidus
In nephrogenic diabetes insipidus (NDI), AVP levels are normal or elevated but the kidney is unable to concentrate urine. The clinical manifestations of polyuria and polydipsia can be present at birth and must be immediately recognised to avoid severe episodes of dehydration. Most (>90%) of congenital patients with NDI have X-linked mutations in the AVPR2 gene, the Xq28 gene coding for the vasopressin V2 (antidiuretic) receptor. In less than 10% of the families studied, congenital NDI has an autosomal recessive inheritance and approximately 42 mutations have been identified in the AQP2 gene (AQP2) located in chromosome region 12q13; that is, the vasopressin-sensitive water channel (fig. 6) [73]. For the AVPR2 gene, 211 putative disease-causing mutations have now been published in 326 unrelated families with X-linked NDI (fig. 7). When studied in vitro, most AVPR2 mutations lead to receptors that are trapped intracellularly and are unable to reach the plasma membrane [74]. A minority of the mutant receptors reach the cell surface but are unable to bind AVP or to trigger an intracellular cAMP signal. Similarly, AQP2 mutant proteins are trapped intracellularly and cannot be expressed at the luminal membrane. This AQP2-trafficking defect is correctable, at least in vitro, by chemical chaperones. Other inherited disorders with mild, moderate, or severe inability to concentrate urine include Bartter syndrome (MIM601678) [75], cystinosis, and autosomal dominant hypocalcaemia [76, 77], nephronophthisis and apparent mineralocorticoid excess [78].
We are recommending the sequencing of the nephrogenic diabetes insipidus genes in all the affected patients. The genes involved are, with a few exceptions, relatively small and easy to sequence. This genomic information is key to the routine care of patients with congenital polyuria and, as in other genetic diseases, reduces health costs and provides psychological benefits to patients and families [79] (fig. 8).
References
1 Ciura S, Liedtke W, Bourque CW. Hypertonicity sensing in organum vasculosum lamina terminalis neurons: a mechanical process involving TRPV1 but not TRPV4. J Neurosci. 2011;31:14669–76.
2 Ramsay DJ, Thrasher TN, Keil LC. The organum vasculosum laminae terminalis: a critical area for osmoreception. Prog Brain Res. 1983;60:91–8.
3 Ciura S, Bourque CW. Transient receptor potential vanilloid 1 is required for intrinsic osmoreception in organum vasculosum lamina terminalis neurons and for normal thirst responses to systemic hyperosmolality. J Neurosci. 2006;26:9069–75.
4 Denton DA, McKinley MJ, Weisinger RS. Hypothalamic integration of body fluid regulation. Proc Natl Acad Sci USA. 1996;93:7397–404.
5 McKinley MJ, Denton DA, Oldfield BJ, De Oliveira LB, Mathai ML. Water intake and the neural correlates of the consciousness of thirst. Semin Nephrol. 2006;26:249–57.
6 Johnson AK. The sensory psychobiology of thirst and salt appetite. Med Sci Sports Exerc. 2007;39:1388–400.
7 Bourque CW. Central mechanisms of osmosensation and systemic osmoregulation. Nat Rev Neurosci. 2008;9:519–31.
8 Hollis JH, McKinley MJ, D’Souza M, Kampe J, Oldfield BJ. The trajectory of sensory pathways from the lamina terminalis to the insular and cingulate cortex: a neuroanatomical framework for the generation of thirst. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1390–401.
9 Lifton RP. Individual genomes on the horizon. N Engl J Med. 2010;362:1235–6.
10 Lechner SG, Markworth S, Poole K, Smith ES, Lapatsina L, Frahm S, et al. The molecular and cellular identity of peripheral osmoreceptors. Neuron. 2011;69:332–44.
11 Leaf A. Nephrology forum: neurogenic diabetes insipidus. Kidney Int. 1979;15:572–80.
12 Babey M, Kopp P, Robertson GL. Familial forms of diabetes insipidus: clinical and molecular characteristics. Nat Rev Endocrinol. 2011;7:701–14.
13 Edwards AM, Mann ME, Marfell-Jones MJ, Rankin DM, Noakes TD, Shillington DP. Influence of moderate dehydration on soccer performance: physiological responses to 45 min of outdoor match-play and the immediate subsequent performance of sport-specific and mental concentration tests. Br J Sports Med. 2007;41:385–91.
14 Saat M, Sirisinghe RG, Singh R, Tochihara Y. Effects of short-term exercise in the heat on thermoregulation, blood parameters, sweat secretion and sweat composition of tropic-dwelling subjects. J Physiol Anthropol Appl Human Sci. 2005;24:541–9.
15 Shirreffs SM, Merson SJ, Fraser SM, Archer DT. The effects of fluid restriction on hydration status and subjective feelings in man. Br J Nutr. 2004;91:951–8.
16 Geelen G, Greenleaf JE, Keil LC. Drinking-induced plasma vasopressin and norepinephrine changes in dehydrated humans. J Clin Endocrinol Metab. 1996;81:2131–5.
17 Andersen LJ, Jensen TU, Bestle MH, Bie P. Gastrointestinal osmoreceptors and renal sodium excretion in humans. Am J Physiol Regul Integr Comp Physiol. 2000;278:R287–94.
18 Claybaugh JR, Sato AK, Crosswhite LK, Hassell LH. Effects of time of day, gender, and menstrual cycle phase on the human response to a water load. Am J Physiol Regul Integr Comp Physiol. 2000;279:R966–73.
19 Verney E. The antidiuretic hormone and the factors which determine its release. Proc R Soc London Ser B. 1947;135:25–6.
20 Zhang Z, Bourque CW. Amplification of transducer gain by angiotensin II-mediated enhancement of cortical actin density in osmosensory neurons. J Neurosci. 2008;28:9536–44.
21 Robertson GL, Athar S. The interaction of blood osmolality and blood volume in regulating plasma vasopressin in man. J Clin Endocrinol Metab. 1976;42:613–20.
22 Sharif Naeini R, Witty MF, Seguela P, Bourque CW. An N-terminal variant of Trpv1 channel is required for osmosensory transduction. Nat Neurosci. 2006;9:93–8.
23 Taylor AC, McCarthy JJ, Stocker SD. Mice lacking the transient receptor vanilloid potential 1 channel display normal thirst responses and central Fos activation to hypernatremia. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1285–1293.
24 McHugh J, Keller NR, Appalsamy M, Thomas SA, Raj SR, Diedrich A, et al. Portal osmopressor mechanism linked to transient receptor potential vanilloid 4 and blood pressure control. Hypertension. 2010;55:1438–43.
25 Boschmann M, Steiniger J, Franke G, Birkenfeld AL, Luft FC, Jordan J. Water drinking induces thermogenesis through osmosensitive mechanisms. J Clin Endocrinol Metab. 2007;92:3334–7.
26 Tank J, Schroeder C, Stoffels M, Diedrich A, Sharma AM, Luft FC, Jordan J. Pressor effect of water drinking in tetraplegic patients may be a spinal reflex. Hypertension. 2003;41:1234–9.
27 Jordan J, Shannon JR, Black BK, Ali Y, Farley M, Costa F, et al. The pressor response to water drinking in humans: a sympathetic reflex? Circulation. 2000;101:504–9.
28 Schroeder C, Bush VE, Norcliffe LJ, Luft FC, Tank J, Jordan J, et al. Water drinking acutely improves orthostatic tolerance in healthy subjects. Circulation. 2002;106:2806–11.
29 Shannon JR, Diedrich A, Biaggioni I, Tank J, Robertson RM, Robertson D, et al. Water drinking as a treatment for orthostatic syndromes. Am J Med. 2002;112:355–60.
30 Claydon VE, Schroeder C, Norcliffe LJ, Jordan J, Hainsworth R. Water drinking improves orthostatic tolerance in patients with posturally related syncope. Clin Sci (Lond) 2006;110:343–52.
31 Stookey JD, Constant F, Popkin BM, Gardner CD. Drinking water is associated with weight loss in overweight dieting women independent of diet and activity. Obesity. (Silver Spring) 2008;16:2481–8.
32 Coste B, Mathur J, Schmidt M, Earley TJ, Ranade S, et al. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science. 2010;330:55–60.
33 Burbach JP. Luckman SM, Murphy D, Gainer H. Gene regulation in the magnocellular hypothalamo-neurohypophysial system. Physiol Rev. 2001;81:1197–267.
34 Rao VV, Loffler C, Battey J, Hansmann I. The human gene for oxytocin-neurophysin I (OXT) is physically mapped to chromosome 20p13 by in situ hybridization. Cell Genet. 1992;61:271–3.
35 Sausville E, Carney D, Battey J. The human vasopressin gene is linked to the oxytocin gene and is selectively expressed in a cultured lung cancer cell line. J Biol Chem. 1985;260:10236–41.
36 Venkatesh B, Si-Hoe SL, Murphy D, Brenner S. Transgenic rats reveal functional conservation of regulatory controls between the Fugu isotocin and rat oxytocin genes. Proc Natl Acad Sci U S A. 1997;94:12462–6.
37 Chen L, Rose JP, Breslow E, Yang D, Chang WR, Furey WFJ, et al. Crystal structure of a bovine neurophysin II dipeptide complex at 2.8 Angström determined from the single-wave length anomalous scattering signal of an incorporated iodine atom. Proc Natl Acad Sci USA. 1991;88:4240–4.
38 Vandesande F, Dierickx K. Identification of the vasopressin producing and of the oxytocin producing neurons in the hypothalamic magnocellular neurosecretory system of the rat. Cell Tissue Res. 1975;164:153–62.
39 Swab D, Pool WC, Novelty F. Immunofluorescence of vasopressin and oxytocin in the rat hypothalamo-neurohypophyseal system. Journal of Neural Transmission. 1975;36:195.
40 Sofroniew M. (1883) Morphology of vasopressin and oxytocin neurones and their central and vascular projections, Vol. 60, Elsevier, New York.
41 Yanovski JA, Friedman TC, Nieman LK, Chrousos GP, Cutler GB, Jr., Doppman JL, et al. Inferior petrosal sinus AVP in patients with Cushing's syndrome. Clin Endocrinol (Oxf) 1997;47:199–206.
42 Kalogeras KT, Nieman LN, Friedman TC, Doppman JL, Cutler GBJ., et al. Inferior petrosal sinus sampling in healthy human subjects reveals a unilateral corticotropin-releasing hormone-induced arginine vasopressin release associated with ipsilateral adrenocorticotropin secretion. J Clin Invest. 1996;97:2045–50.
43 Donaldson ZR, Young LJ. Oxytocin, vasopressin, and the neurogenetics of sociality. Science. 2008;322:900–4.
44 Serradeil-Le Gal C, Wagnon J, Simiand J, Griebel G, Lacour C, Guillon G, et al. Characterization of (2S,4R)-1-[5-chloro-1-[(2,4-dimethoxyphenyl)sulfonyl]-3-(2-methoxy-phenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-4-hydroxy-N,N-dimethyl-2-pyrrolidine carboxamide (SSR149415), a selective and orally active vasopressin V1b receptor antagonist. J Pharmacol Exp Ther. 2002;300:1122–30.
45 Thibonnier M, Coles P, Thibonnier A, Shoham M. The basic and clinical pharmacology of nonpeptide vasopressin receptor antagonists. Annu Rev Pharmacol Toxicol. 2001;41:175–202.
46 Walum H, Westberg L, Henningsson S, Neiderhiser JM, Reiss D, Igl W, et al. Genetic variation in the vasopressin receptor 1a gene (AVPR1A) associates with pair-bonding behavior in humans. Proc Natl Acad Sci U S A. 2008;105:14153–6.
47 Serradeil-Le Gal C, Wagnon J, 3rd, Tonnerre B, Roux R, Garcia G, Griebel G, and Aulombard A. An overview of SSR149415, a selective nonpeptide vasopressin V(1b) receptor antagonist for the treatment of stress-related disorders. CNS Drug Rev. 2005;11:53–68.
48 Seibold A, Brabet P, Rosenthal W, Birnbaumer M. Structure and chromosomal localization of the human antidiuretic hormone receptor gene. Am J Hum Genet. 1992;51:1078–83.
49 Birnbaumer M, Seibold A, Gilbert S, Ishido M, Barberis C, Antaramian A, et al. Molecular cloning of the receptor for human antidiuretic hormone. Nature. 1992;357:333–5.
50 Fredriksson R, Lagerstrom MC, Lundin LG, Schioth HB. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol. 2003;63:1256–72.
51 Perez DM. The evolutionarily triumphant G-protein-coupled receptor. Mol Pharmacol. 2003;63:1202–5.
52 Nielsen S, Frokiaer J, Marples D, Kwon TH, Agre P, Knepper MA. Aquaporins in the kidney: from molecules to medicine. Physiol Rev. 2002;82:205–44.
53 Boone M, Deen PM. Physiology and pathophysiology of the vasopressin-regulated renal water reabsorption. Pflugers Arch. 2008;456:1005–24.
54 Nedvetsky PI, Tamma G, Beulshausen S, Valenti G, Rosenthal W, Klussmann E. Regulation of aquaporin-2 trafficking. Handb Exp Pharmacol. 2009;133–57.
55 Smith CP. Mammalian urea transporters. Exp Physiol. 2009;94:180–5.
56 Yang B, Bankir L, Gillespie A, Epstein CJ, Verkman AS. Urea-selective concentrating defect in transgenic mice lacking urea transporter UT-B. J Biol Chem. 2002;277:10633–7.
57 Bankir L, Fernandes S, Bardoux P, Bouby N, Bichet DG. Vasopressin-V2 receptor stimulation reduces sodium excretion in healthy humans. J Am Soc Nephrol. 2005;16:1920–8.
58 Morishita T, Tsutsui M, Shimokawa H, Sabanai K, Tasaki H, Suda O, et al. Nephrogenic diabetes insipidus in mice lacking all nitric oxide synthase isoforms. Proc Natl Acad Sci U S A. 2005;102:10616–21.
59 Olesen ET, Rutzler MR, Moeller HB, Praetorius HA, Fenton RA. Vasopressin-independent targeting of aquaporin-2 by selective E-prostanoid receptor agonists alleviates nephrogenic diabetes insipidus. Proc Natl Acad Sci U S A. 2011;108:12949–54.
60 Sands JM, Naruse M, Baum M, Jo I, Hebert SC, Brown EM, Harris HW. Apical extracellular calcium/polyvalent cation-sensing receptor regulates vasopressin-elicited water permeability in rat kidney inner medullary collecting duct. J Clin Invest. 1997;99:1399–405.
61 McKusick VA. (2000) Online Mendelian Inheritance in Man OMIM, (TM). McKusick-Nathans Institute for Genetic Medicine, Johns Hopkins University (Baltimore, MD) and National Center for Biotechnology Information, National Library of Medicine (Bethesda, MD), 2000. World Wide Web URL: http://www.ncbi.nlm.nih.gov/omim/.
62 Rittig R, Robertson GL, Siggaard C, Kovacs L, Gregersen N, Nyborg J, Pedersen EB. Identification of 13 new mutations in the vasopressin-neurophysin II gene in 17 kindreds with familial autosomal dominant neurohypophyseal diabetes insipidus. Am J Hum Genet. 1996;58:107–17.
63 Brachet C, Birk J, Christophe C, Tenoutasse S, Velkeniers B, et al. Growth retardation in untreated autosomal dominant familial neurohypophyseal diabetes insipidus caused by one recurring and two novel mutations in the vasopressin-neurophysin II gene. Eur J Endocrinol. 2011;164:179–87.
64 Birk J, Friberg MA, Prescianotto-Baschong C, Spiess M, Rutishauser J. Dominant pro-vasopressin mutants that cause diabetes insipidus form disulfide-linked fibrillar aggregates in the endoplasmic reticulum. J Cell Sci. 2009;122:3994–4002.
65 Castino R. Davies J, Beaucourt S, Isidoro C, Murphy D. Autophagy is a prosurvival mechanism in cells expressing an autosomal dominant familial neurohypophyseal diabetes insipidus mutant vasopressin transgene. FASEB J. 2005;19:1021–3.
66 Abu Libdeh A, Levy-Khademi F, Abdulhadi-Atwan M, Bosin E, Korner M, White PC, et al. Autosomal recessive familial neurohypophyseal diabetes insipidus: onset in early infancy. Eur J Endocrinol. 2009;162:221–6.
67 Willcutts MD, Felner E, White PC. Autosomal recessive familial neurohypophyseal diabetes insipidus with continued secretion of mutant weakly active vasopressin. Hum Mol Genet. 1999;8:1303–7.
68 Christensen JH, Kvistgaaard H, Knudsen J, Shaikh G, Tolmie J, Cooke S, et al. A novel deletion partly removing the Avp gene causes autosomal recessive inheritance of early onset neurohypophyseal diabetes insipidus. Clin Genet, (2011) in press.
69 Bichet DG, Arthus M-F, Lonergan M, Morgan K, Fujiwara TM. Hereditary central diabetes insipidus: autosomal dominant and autosomal recessive phenotypes due to mutations in the prepro-AVP-NPII gene. J Am Soc Nephrol. 1998;9:386A.
70 Christensen JH, Siggaard C, Corydon TJ, Robertson GL, Gregersen N, Bolund L, et al. Differential cellular handling of defective arginine vasopressin (AVP) prohormones in cells expressing mutations of the AVP gene associated with autosomal dominant and recessive familial neurohypophyseal diabetes insipidus. J Clin Endocrinol Metab. 2004;89:4521–31.
71 Beuret N, Stettler H, Renold A, Rutishauser J, Spiess M. Expression of regulated secretory proteins is sufficient to generate granule-like structures in constitutively secreting cells. J Biol Chem. 2004;279:20242–9.
72 Tian W, Fu Y, Garcia-Elias A, Fernandez-Fernandez JM, Vicente R, Kramer PL, et al. A loss-of-function nonsynonymous polymorphism in the osmoregulatory TRPV4 gene is associated with human hyponatremia. Proc Natl Acad Sci U S A. 2009;106:14034–9.
73 Robben JH, Knoers NV, Deen PM. Cell biological aspects of the vasopressin type-2 receptor and aquaporin 2 water channel in nephrogenic diabetes insipidus. Am J Physiol Renal Physiol. 2006;291:F257–270.
74 Spanakis E, Milord E, Gragnoli C. AVPR2 variants and mutations in nephrogenic diabetes insipidus: review and missense mutation significance. J Cell Physiol. 2008;217:605–17.
75 Peters M, Jeck N, Reinalter S, Leonhardt A, Tonshoff B, Klaus GG, et al. Clinical presentation of genetically defined patients with hypokalemic salt-losing tubulopathies. Am J Med. 2002;112:183–90.
76 Konrad M, Weber S. Recent advances in molecular genetics of hereditary magnesium-losing disorders. J Am Soc Nephrol. 2003;14:249–60.
77 Gahl WA, Thoene JG, Schneider JA. Cystinosis. N Engl J Med. 2002;347:111–21.
78 Bockenhauer D, van’t Hoff W, Dattani M, Lehnhardt A, Subtirelu M, Hildebrandt F, et al. Secondary nephrogenic diabetes insipidus as a complication of inherited renal diseases. Nephron Physiol. 2010;116:23–9.
79 Green ED, Guyer MS. Charting a course for genomic medicine from base pairs to bedside. Nature. 2011;470:204–13.
80 Wilson Y, Nag N, Davern P, Oldfield BJ, McKinley MJ, Greferath U, Murphy M. Visualization of functionally activated circuitry in the brain. Proc Natl Acad Sci U S A. 2002;99:3252–7.
81 Prager-Khoutorsky M, Bourque CW. Osmosensation in vasopressin neurons: changing actin density to optimize function. Trends Neurosci. 2010;33:76–83.
82 Deen PMT, Verdijk MAJ, Knoers NVAM, Wieringa B, Monnens LAH, van Os CH, et al. Requirement of human renal water channel aquaporin-2 for vasopressin-dependent concentration of urine. Science. 1994;264:92–5.