The future of osteoporosis treatment – a research update
DOI: https://doi.org/10.4414/smw.2012.13624
Summary
Osteoporosis is characterised by a progressive loss of bone mass and microarchitecture which leads to increased fracture risk. Some of the drugs available to date have shown reductions in vertebral and non-vertebral fracture risk. However, in the ageing population of industrialised countries, still more fractures happen today than are avoided, which highlights the large medical need for new treatment options, models, and strategies. Recent insights into bone biology, have led to a better understanding of bone cell functions and crosstalk between osteoblasts, osteoclasts, and osteocytes at the molecular level. In the future, the armamentarium against osteoporotic fractures will likely be enriched by (1.) new bone anabolic substances such as antibodies directed against the endogenous inhibitors of bone formation sclerostin and dickkopf-1, PTH and PTHrp analogues, and possibly calcilytics; (2.) new inhibitors of bone resorption such as cathepsin K inhibitors which may suppress osteoclast function without impairing osteoclast viability and thus maintain bone formation by preserving the osteoclast-osteoblast crosstalk, and denosumab, an already widely available antibody against RANKL which inhibits osteoclast formation, function, and survival; and (3.) new therapeutic strategies based on an extended understanding of the pathophysiology of osteoporosis which may include sequential therapies with two or more bone active substances aimed at optimising the management of bone capital acquired during adolescence and maintained during adulthood in terms of both quantity and quality. Finally, one of the future challenges will be to identify those patients and patient populations expected to benefit the most from a given drug therapy or regimen. The WHO fracture risk assessment tool FRAX® and improved access to bone mineral density measurements by DXA will play a key role in this regard.
The medical need for new osteoporosis drugs
The World Health Organization (WHO) defined osteoporosis as “a systemic skeletal disease characterised by low bone mass and microarchitectural deterioration of bone tissue, with a consequent increase in bone fragility and susceptibility to fracture” [1]. With the ageing of the population, the complications of osteoporosis, fractures, represent a growing medical and socio-economic threat in industrialised countries. Switzerland belongs to the countries with the highest and still fastest growing life expectancy at birth worldwide (84.6 years for women and 80.2 years for men in 2010, corresponding to +2.0 and +3.3 years gained since 2000) [2]. The recent demographic scenarios projecting the aging of the population in Switzerland between 2005 and 2050 showed that this increasing trend will not level-off before year 2050, leading to almost a doubling of the population older than 65 years of age by that date [3]. As the incidence of osteoporotic fractures increases exponentially with age, the implementation of measures aimed at reducing fracture risk is needed to preserve quality of life and to ensure adequate control of health care costs. It has been shown that the direct medical costs of hospitalisations for osteoporotic fractures in women and men already exceeded today the costs of hospitalisations of many other chronic diseases, such as major cardiovascular events (stroke, myocardial infarction, heart failure), breast cancer, and chronic obstructive pulmonary diseases [4].
The goal of osteoporosis treatment is to reduce fractures. This can be achieved either by decreasing bone resorption and/or by increasing bone formation.
Bisphosphonates are today’s mainstay of osteoporosis treatment. They act as inhibitors of bone resorption with a high affinity for bone and were shown to increase BMD and reduce fracture risk in patients with postmenopausal, male, and glucocorticosteroid-induced osteoporosis. Due to their (very) long terminal half-life in bone, they can be administered either orally (daily, weekly, or monthly tablets) exposing the patient to a risk of esophageal irritation or intravenously (quarterly or yearly infusions), exposing the patient to the risk of an acute-phase reaction. In addition, intravenous bisphosphonates are contra-indicated in patients with restricted renal function. The bisphosphonates alendronate, risedronate, and zoledronate were shown to reduce the risk of new vertebral, non-vertebral, and hip fractures [5–9]. However, while relative fracture risk reduction reached 50% for spine and hip fractures, the order of magnitude for non-vertebral fracture risk reduction was around 20%. While long term treatment of osteoporosis with a bisphosphonate up to 10 years has shown excellent safety [10], bisphosphonate therapy has been associated with a potential risk of osteonecrosis of the jaw and of atypical subtrochanteric femoral fractures [11–13]. Other available drugs that reduce bone resorption are the oral selective estrogen receptor modulators (SERMs) raloxifene and basedoxifene, and subcutaneous denosumab, a human monoclonal antibody that inhibits RANKL. Raloxifene was shown to reduce vertebral fracture and breast cancer risk in postmenopausal women [14–17]. SERMs increase the risk of venous thromboembolic events, may increase the risk [16] of fatal stroke in postmenopausal women at increased coronary risk, and may trigger or aggravate climacteric symptoms in younger postmenopausal women [15, 16]. Finally, SERMs are not indicated in men. In postmenopausal women with osteoporosis, the bone resorption inhibitor denosumab reduced vertebral, non-vertebral and hip fracture risk by the same order of magnitude as bisphosphonates (–68%, –20%, and –40%, respectively) [18]. The incidence of cancers and infections was comparable to that of placebo in controlled trials. However, due to the tight interrelationship with shared signaling pathways between the skeletal and immune systems, potentially associated risks deserve further scrutiny [19, 20]. Osteonecrosis of the jaw was not seen in the pivotal trial but was reported in the extension study [21, 22] and in studies with patients with metastatic bone disease or myeloma [23, 24]. Compared to bisphosphonates, denosumab exhibits a much shorter terminal half-life of several months instead of several years [25].
In women with postmenopausal osteoporosis, daily subcutaneous injections of full length parathormone (1-84 PTH) and teriparatide (the 1-34 N-terminal fragment of PTH) showed the largest increases in BMD reported to date for any treatment of osteoporosis, accompanied by an important reduction of the risk of new vertebral fractures [26, 27]. Teriparatide was shown to reduce the risk of vertebral and non-vertebral but not hip fractures in postmenopausal and glucocorticoid-induced osteoporosis [27, 28]. Although the safety and efficacy of teriparatide have been studied beyond two years of treatment, the generally approved duration of therapy is limited to two years.
In summary, osteoporosis and its complications, the fragility fractures, belong to the leading chronic diseases in industrialised countries imposing a growing burden to patients, society and healthcare systems. Treatments available to date are imperfect:
– While therapeutic alternatives are available for inhibiting bone resorption, options are much more limited with regard to bone anabolic substances. There is a need for new bone active substances aimed at restoring quantitatively and qualitatively normal bone.
– While the safety profile of most bone active substances is at least partially known, there is considerable potential for improvement (ONJ, atypical fractures, interactions with the immune system). Treatment alternatives are needed for patients exhibiting or at risk of side-effects.
– While a relative fracture risk reduction of up to 20%, 50%, and 70% can be achieved for non-vertebral, hip, and spine fractures, respectively, still 30 to 80% of these fractures cannot be prevented. As osteoporosis is a systemic disease, there is an obvious need for improving the magnitude of the therapeutic effect at all fracture sites.
– While patients included in pivotal randomised controlled trials usually had osteoporosis defined according to the WHO criteria, i.e. a T-score below –2.5 SD and / or prevalent fragility fractures, a large proportion of fractures occurs at T-scores above –2.5 SD and in patients without prior fractures. Thus, therapies with proven fracture risk reduction efficacy in patients at increased fracture risk, e.g. in patients with osteopenia and/or clinical risk factors, might contribute to earlier and more efficient intervention against fractures.
– While fracture risk increases with age, most studies were performed in patients below 80 years of age. Osteoporosis drugs with a good safety profile and proven to reduce fracture risk in the elderly and the very old are lacking.
For all these reasons, there is an urgent need for the development of new bone active substances aimed at treating osteoporosis and preventing fractures.
New insights in bone biology
Bone remodeling is the continuous process by which old bone is removed by bone-resorbing cells, the osteoclasts, and replaced by new bone synthesised by bone forming cells, the osteoblasts. During adult life bone remodeling primarily serves the purposes of regulating calcium homeostasis and repairing bone micro-fractures resulting from mechanical loading. Knowing that the skeleton is completely remodeled every ten years, microfracture repair prevents its excessive ageing by preventing the accumulation of old bone [29]. At their smallest functional level, the bone remodeling unit, osteoblasts and osteoclasts are tightly coupled by paracrine signaling whereby the net balance in new bone built may be positive (during growth), neutral (at peak bone mass) or negative (in osteoporosis and other diseases leading to a net loss of bone mass). Several molecules that either promote or inhibit the activity of bone remodeling and its constituent cells have been recently identified, giving new insights into bone pathophysiology and opening the gateway for possible future therapeutic options.
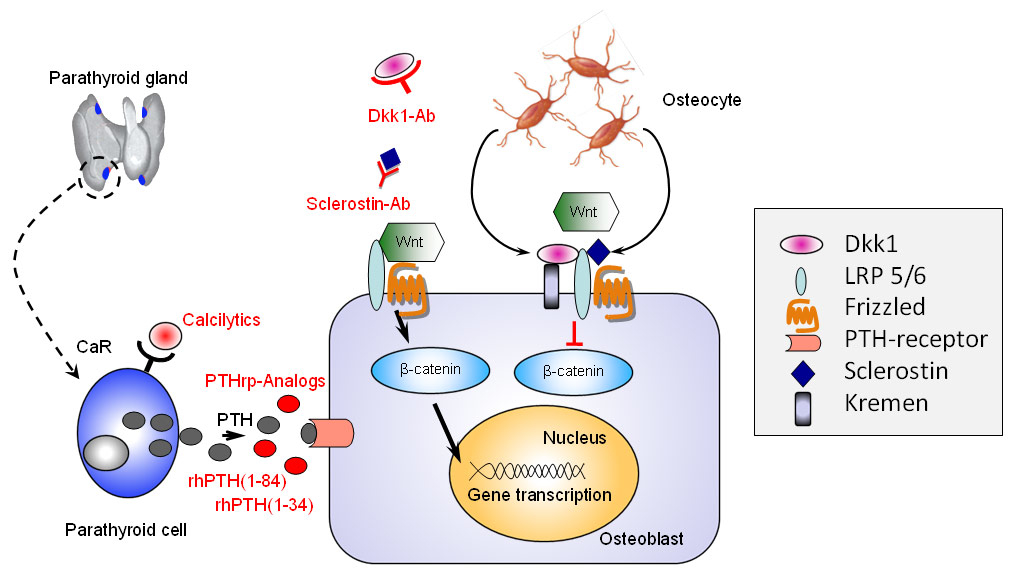
Figure 1
Targeting osteoblasts to stimulate bone formation.
Recombinant human full length parathormone, rhPTH (1-84), its N-terminal segment rhPTH (1-34), and PTH related protein (PTHrp) analogues exert their bone anabolic effect after binding to the PTH receptor present on the osteoblasts. Calcilytics modulate PTH secretion by shifting the setpoint of the calcium-receptor (CaR) found on parathyroid cells. Sclerostin and Dickkopf-1 (dkk1) are endogenous inhibitors of the canonical Wnt-ß-catenin signaling pathway. Monoclonal antibodies directed against sclerostin or dkk1 induce an intracellular accumulation of ß-catenin followed by increased gene transcription which ultimately results into stimulation of osteoblastic bone formation.
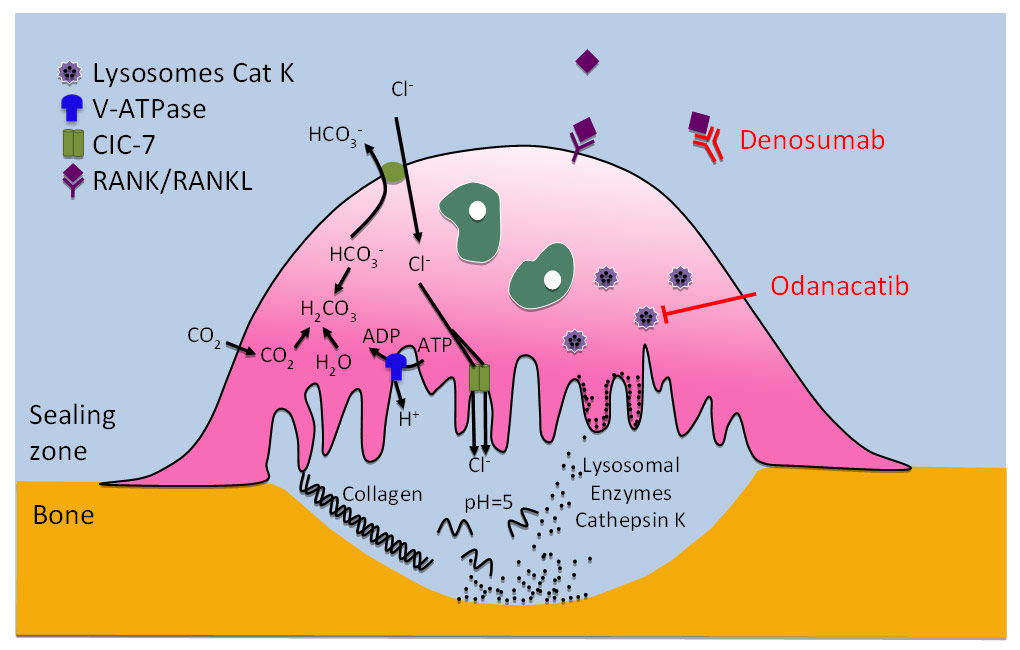
Figure 2
Targeting osteoclasts to inhibit bone resorption.
By binding to the RANK-ligand (RANKL), the recombinant human IgG2-antibody denosumab prevents the interaction of RANKL with its receptor RANK, which leads to the inhibition of osteoclast formation, function, and survival, and to decreasing bone resorption. Odanacatib is an inhibitor of cathepsin K, a lysosomal cysteine protease with high collagenase activity expressed predominantly in osteoclasts, which may suppress osteoclast function without impairing osteoclast viability, thus preserving the osteoclast-osteoblast crosstalk and bone formation.
Osteoblasts
The osteoblasts are derived from mesenchymal stem cells, produce the bone matrix which consists of collagenous and non-collagenous proteins (such as type I collagen and osteocalcin, respectively), and control its subsequent mineralisation, that is the deposition of hydroxyapatite. Thus, the rate of bone formation will depend upon individual osteoblast activity, their lifespan and the number of precursor cells recruited. As shown in figure 1, parathormone (PTH) and PTH related protein (PTHrp), the only other known ligand of the PTH receptor, increase the lifespan of mature osteoblasts by reducing their rate of apoptosis, which explains their anabolic effects on bone [30]. The calcium-sensing receptor (CaR) which is located on the surface of the parathyroid gland senses extracellular levels of ionised calcium and controls calcium homeostasis by regulating the release of PTH. Calcimimetics mimic the effects of ionized calcium on the CaR, create a leftward shift in the set point for PTH secretion, i.e. decrease the secretion of PTH [31, 32]. Calcilytics do the opposite, that is mimic low ionised concentrations on the CaR which induces PTH secretion by the parathyroid glands [32].
As recently discovered, osteoblastic differentiation and the number of osteoblast precursor cells recruited is regulated by a core molecular pathway, the canonical Wnt/β-catenin pathway [33]. Wnt, the acronym resulting from a combination of two genes found in Drosophila (“Wingless”) and vertebrates (“Int”), stands for a specific family of proteins involved in the Wnt signaling pathway. The latter plays diverse roles ranging from embryology (e.g., during mesoderm, neurectoderm and body axis formation), and bone modeling and remodeling, to causing diseases such as cancer. When activated by Wnt, the pathway ultimately leads to the accumulation of β–catenin in the nucleus and to the subsequent gene transcription, i.e., to signal-dependent cell differentiation. When a cell is not exposed to a Wnt signal, β–catenin is degraded and gene transcription is stopped [34–36]. In bone, the Wnt signaling pathway plays a key role in osteoblast differentiation so that agents directed against endogenous Wnt inhibitors specific to bone, might selectively permit accelerated osteoblast differentiation and thus increase the bone formation rate [33, 36].
Osteocytes
During the process of matrix building and mineralisation, some osteoblasts remain trapped within lacunae and are named osteocytes, characterised by their dendritic extensions in the bone canaliculi which, in analogy with the dendritic network of the nervous system, allow them to communicate with each other and with cells on the bone surface. Although osteocytes are the most common cell type in bone, they were neglected by research for many years, possibly due to their discrete level of activity. However, osteocytes were shown to play a key role in the regulation of phosphate metabolism (as the source of FGF-23) and to be involved in sensing and transferring information possibly by acting as the not yet fully understood but long sought mechanosensor controlling bone remodeling [37]. As shown in figure 1, Osteocytes were shown to be almost the only source of the endogenous Wnt-inhibitor sclerostin [38] which may be one of the inhibitory signals they use to transmit mechanical load information to the effector cells [39–41].
Osteoclasts
The osteoclasts are issued from haemopoietic stem cells and are formed by the fusion of cells from the monocyte-macrophage cell line. Osteoclastogenesis requires the presence of RANKL (Receptor Activator of Nuclear factor κβ Ligand), a member of the TNF (Tumor Necrosis Factor) cytokines, and M-CSF (Macrophage-Colony Stimulating Factor). Once RANKL is expressed by the osteoblasts, it activates its receptor RANK on the cells of the osteoclast lineage leading to their proliferation, maturation, activation, and survival, ultimately resulting into increased bone resorption. Osteoprotegerin (OPG), a soluble decoy receptor of RANKL also produced by the osteoblasts, acts as its natural antagonist [42]. Thus, as shown in figure 2, an antibody directed against RANKL, such as denosumab, will mimic the effects of OPG and reduce the production, maturation, and activity of osteoclasts by reversibly hampering the RANK/RANKL interaction, ultimately leading to the inhibition of osteoclast-mediated bone resorption [43]. RANKL and its receptor RANK are also expressed on T lymphocytes and antigen-presenting dendritic cells, suggesting the existence of a crosstalk between immune cells and bone. The latter provides a rationale for the bone loss observed in patients with a chronically activated immune system such as in rheumatoid arthritis or leukaemias [43]. Furthermore, the RANKL-RANK-OPG system is involved in other processes such as in controlling autoimmunity or immune responses in the skin [43].
Once the osteoclasts are attached and sealed to bone surface, they produce an acid medium which dissolves the mineral component, leaving the organic matrix exposed to the lytic effects of enzymes such as cysteine protein kinases [44]. With regard to bone remodeling, cathepsin K is the most important of these enzymes since it has exceptionally high collagenase activity towards triple helical collagens in an acidic environment [45]. While cathepsin K is expressed predominantly in osteoclasts and various other multinucleated cells such as foreign-body giant cells and Langhans cells, it is also found to a lesser degree in macrophages, synovial fibroblasts, and fibroblasts at locations of wound healing or inflammation, chondrocytes, various epithelial cells of the human fetus, adult lung airway epithelium, thyroid epithelium, and possibly at low concentrations in smooth muscle cells [45]. The natural model of human pycnodysostosis, an autosomal recessive bone dysplasia caused by cathepsin K deficiency [46] and the observation of osteopetrosis in cathepsin K-deficient mice [47] underlines the key role played by cathepsin K in bone resorption and the potential of selective cathepsin K inhibitors as future bone resorption inhibitors for therapeutic use.
New and emerging treatment options of osteoporosis
These recent insights into bone pathophysiology and the better understanding of the core mechanisms involved in the development of osteoporosis have identified new targets for intervention and led to the development of molecules with therapeutic potential.
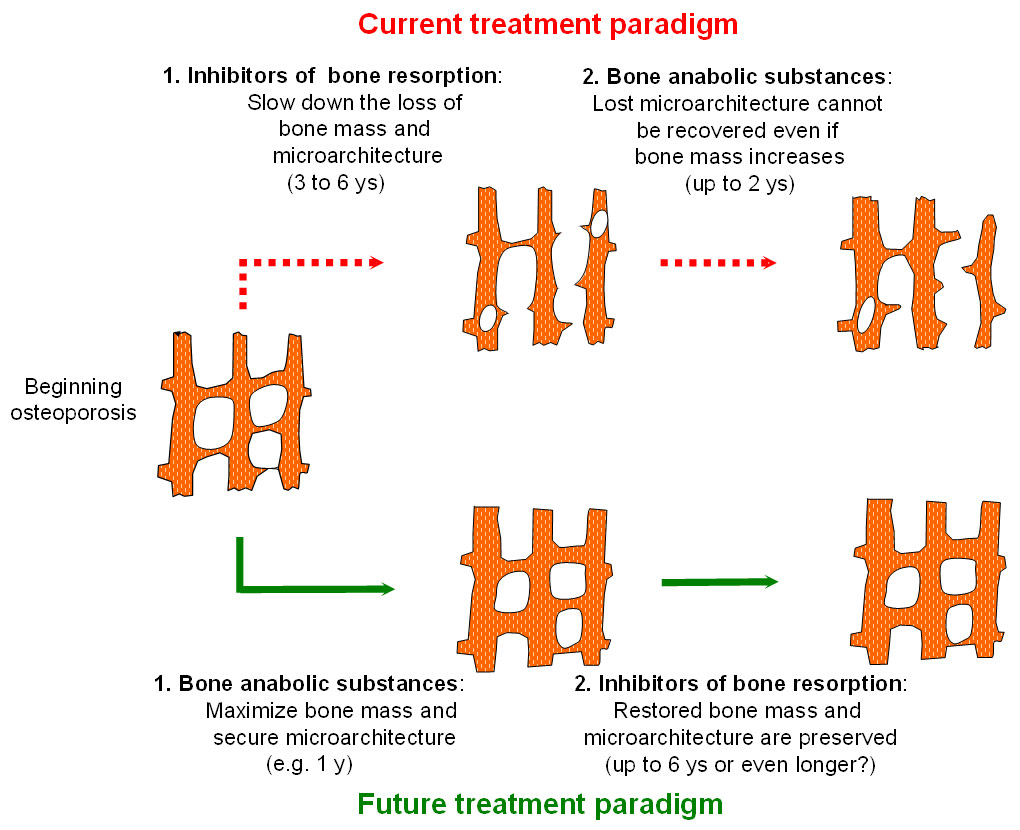
Figure 3
Current and future treatment paradigm of osteoporotic bone loss of bone mass and microarchitecture.
According to the current treatment paradigm, antiresorptive drugs are the mainstay first line therapy for reducing fracture risk in patients with osteoporosis and anabolic substances are usually recommended in patients with severe osteoporosis only. As lost bone microarchitecture cannot be recovered, a future treatment paradigm may aim at reversing osteoporosis by maximising bone mass and preserving bone microarchitecture during the early phase of disease progression, the achieved gains being preserved by an inhibitor of bone resorption used in a second step.
Stimulation of bone formation
PTH and PTHrp analogues
In many countries, the only available bone anabolic substance is human recombinant N-terminal 1-34 PTH (teriparatide). Full length 1-84 PTH is available in Europe but has not yet been approved in the US and in Switzerland. Due to its unique efficacy profile, a broadened use of teriparatide as a first line treatment for postmenopausal women and for men with severe osteoporosis has been recently advocated [48]. However, there is a need for accessible and more convenient bone anabolic agents. A teriparatide-coated microneedle patch system (ZP-PTH) was developed and the results of a phase II dose-finding study showed an increase in biochemical markers of bone turnover and in lumbar spine BMD [49, 50].
PTHrp is a protein analogue to 1-84 PTH which has physiological functions, such as the stimulation of bone resorption allowing tooth eruption and the control of calcium metabolism in lactating women [51]. PTHrp also plays an important paraneoplastic role in hypercalcaemia of malignancy [52]. While the continuous administration of PTH leads to bone resorption over formation, i.e., to a net loss of bone mass, the continuous administration of PTHrp preferentially stimulates bone formation. It was recently suggested that this apparently discrepant finding may be due to the existence of two activated states of the PTH receptor with continuous cAMP secretion after PTH binding and burst cAMP secretion after PTHrp binding [53], thus mimicking the effects of intermittently administered PTH, such as reduced osteoblast apoptosis [54]. A randomised, placebo- and teriparatide- controlled dose-finding study with the PTHrp analogue BA-058 in 222 women with postmenopausal osteoporosis (mean age 65 years) was completed in 2009, showing a dose-dependent increase in BMD at the spine, the total hip and the femoral neck after 6 months which was comparable to (or at the highest dose (SC 80 µd/d) even numerically higher than) that achieved with teriparatide with comparable tolerability (NCT00542425). A randomised controlled phase III trial comparing the effects of BA-058 80 µd/d SC with teriparatide 20 µd/d SC and SC placebo on the number of new morphometric vertebral fractures after 18 months in postmenopausal women with osteoporosis is currently ongoing (NCT01343004).
Calcilytics
Oral CaR-antagonists or calcilytics, such as JTT-305/MK-5442 and SB-423557, were shown to stimulate endogenous transient PTH secretion and bone formation and to prevent bone loss in ovariectomised rats, thus delivering a proof of concept [55, 56]. However, after two dose-finding studies in women with postmenopausal osteoporosis (NCT00996801 and NCT00960934), the development of MK-5442, the most advanced in development, was halted in 2011 for unreported reasons.
Antagonists of Wnt-inhibitors
Sclerostin (SOST) and dickkopf-1 (dkk1) are endogenous inhibitors of the canonical Wnt/β–catenin pathway specific to bone. In the presence of these inhibitors, the osteoblast precursors are not exposed to a Wnt signal, so that β–catenin is degraded and osteoblast differentiation and recruitment is stopped. Antibodies directed against sclerostin or dickkopf-1, should therefore have bone anabolic properties.
Sclerosteosis is a rare autosomal recessive craniotubular hyperostosis caused by one of at least six known inactivating mutations of the single gene which encodes for sclerostin [57–59]. Homozygote carriers are exposed to facial palsy and deafness during childhood, as a result of cranial nerve entrapment, and to development of elevated intracranial pressure, as a result of calvarial overgrowth. The complications of the latter are usually the cause of death [60, 61]. Patients with sclerosteosis have bone of normal quality [62]. Heterozygous carriers of sclerosteosis have BMD values consistently higher than the mean of healthy subjects without any of the bone complications encountered in homozygotes [63]. Sclerosteosis and the closely related Van Buchem disease [64–66] represent a natural SOST gene knock-out model in humans, which was seminal for the development of sclerostin antibodies as anabolic bone substances. In preclinical studies, the administration of sclerostin-neutralising monoclonal antibodies was shown to increase bone formation on trabecular, periosteal, endocortical, and intracortical surfaces without increasing bone resorption (as seen with teriparatide or full length PTH), and to increase trabecular thickness, BMD and bone strength [67, 68]. The subcutaneous administration of a single dose of AMG 785, a human recombinant sclerostin antibody, to healthy men and postmenopausal women showed dose-related increases in bone formation markers with a dose-related decrease in one bone resorption marker (serum C-telopeptide, CTx), suggesting a large anabolic window. In addition, BMD increases at the lumbar spine and the hip were reported on day 85 after the single injection. AMG 785 was generally well tolerated. One treatment-related serious adverse event of nonspecific hepatitis was reported and six subjects in the higher-dose groups developed anti-AMG 785 antibodies, 2 of which were neutralising [69]. A dose-finding randomised placebo-, teriparatide- and alendronate-controlled 12-month phase II study in postmenopausal women with low BMD was recently completed (NCT00896532). According to a company press release available under http://www.amgen.com , “significant increases in lumbar spine bone mineral density at month 12 versus the placebo arm” were demonstrated, and “AMG 785 compared positively with the two active comparators, teriparatide and alendronate.” Furthermore, “the overall incidence of adverse events was generally balanced between groups. Consistent with previous studies, injection site reactions were reported more frequently in those patients receiving AMG 785”. Two additional phase II fracture healing studies with AMG 785 (in tibial diaphyseal fractures post intramedullary nailing [NCT00907296] and in low energy intertrochanteric or femoral neck fractures [NCT01081678]) are currently ongoing. Recruitement for phase III studies in post-menopausal osteoporosis has recently begun. Provided that sclerostin antibodies will be shown to reduce fracture risk with a good safety and tolerability profile, the perspective of uncoupling bone formation and resorption and of restoring bone microarchitecture would represent a quantum leap in the future management of osteoporosis.
Dickkopf-1 is a protein encoded by the dkk1 gene which is associated with the presence of osteolytic lesions in patients with multiple myeloma [70]. Serum dickkopf-1 concentrations are significantly higher in patients with low BMD and in women with postmenopausal osteoporosis [71, 72] and the waning effect of teriparatide treatment on bone turnover over time was shown to be associated with an increase in serum dickkopf-1 [72, 73]. Thus, it would be theoretically possible to develop antibodies specifically directed against dickkopf-1 which might have anabolic effects of bone.
The availability of new anabolic substances might contribute to a paradigm shift with regard to the sequence in which anti-osteoporosis drugs will be administered in the future. According to current practice as well as to international recommendations and guidelines, antiresorptive drugs, such as bisphosphonates, are the mainstay first line therapy for reducing fracture risk in patients with osteoporosis. In patients with severe osteoporosis, some of which may fracture during therapy with a bone resorption inhibitor, anabolic substances are recommended [74–76]. However, as shown in figure 3, the reverse sequence, i.e., starting with a bone anabolic substance at early stages of osteoporosis will contribute to increasing bone mass and keeping bone microarchitecture intact, as much as possible. Thereafter, preserving newly acquired bone mass and intact microarchitecture should allow for long term and extended fracture risk prevention. Clinical trials aimed at documenting the efficacy and safety of such reverse treatment sequences are currently underway (NCT01575834).
It should be remembered that mutations within the Wnt signaling pathway have been associated with many forms of cancer, including but not limited to gastric, kidney, liver, lung, ovarian, and bone cancer [36, 77]. Studies in human osteosarcoma cell lines have shown activation of Wnt signaling with loss of Wnt inhibitory factor 1, suggesting that sclerostin inhibition may increase susceptibility to osteosarcoma [78]. Although patients with osteosclerosis, do not appear to be exposed to increased cancer risk, their considerably shortened average life expectancy does not allow firm conclusions. It should also be remembered that the Wnt signaling pathway is cell specific and that its modulation is not an on-off situation. Conceptually, at its lowest level of activity, Wnt-signaling may contribute to osteoporosis, at a medium level to normal bone physiology, at a higher level to increased bone mass and at its highest level to the development of cancer. Thus, the research challenge will be to identify specific antibodies directed against Wnt-modulators specific to bone cells and to achieve a dose-response within a to be defined therapeutic non carcinogenetic range [36].
Inhibition of bone resorption
RANKL-inhibitors
Denosumab, the first in class RANKL-inhibitor, is a recombinant human IgG2 antibody with affinity and specificity for RANKL. By binding to RANKL, denosumab prevents the RANKL/RANK interaction on the osteoblast which leads to the inhibition of osteoclast formation, function, and survival, thereby decreasing bone resorption and increasing bone mass and strength in both cortical and trabecular bone. The 3-year, randomised, double-blind, placebo-controlled fracture endpoint trial FREEDOM enrolled 7808 women between the ages of 60 and 90 years (mean 72 years) who had a baseline BMD T-score between –2.5 and –4.0 at either the lumbar spine or total hip. The mean baseline lumbar spine BMD T-score was –2.8 SD, and 23% of women had a vertebral fracture at baseline. Women were randomised to receive SC injections of either placebo (N = 3906) or denosumab 60 mg (N = 3902) once every 6 months. All women received at least 1000 mg calcium and 400 IU vitamin D supplementation daily. Denosumab significantly reduced the incidence of new morphometric vertebral fractures (primary endpoint) at 3 years (7.2% vs 2.3%, –68%, p <0.0001). In addition, denosumab significantly reduced the incidence of hip and non-vertebral fractures (secondary endpoints) at 3 years (1.2% vs 0.7%, –40%, p = 0.04 and 8.0% vs 6.5%, –20%, p = 0.01, respectively) [18]. Furthermore, the antifracture efficacy of denosumab was consistent across patients with varying degrees of fracture risk [79]. Denosumab was generally well tolerated. Based on postmarketing and clinical research experience available to date, denosumab exposes patients to a risk of hypocalcaemia, which is significant in patients with severe renal impairment or receiving dialysis, to a potential for adverse outcomes resulting from the induced profound, even if reversible, suppression of bone remodelling such as osteonecrosis of the jaw, atypical fractures and delayed fracture healing, and to a potentially increased risk of severe infections consistent with its osteoimmunological effects. Epidermal and dermal adverse events not specific to the injection site (such as dermatitis, eczema, and rashes) were significantly increased. Finally, cases of pancreatitis and new malignancies of the breast, the reproductive system, and the gastrointestinal system were numerically more frequent with denosumab with no established causal relationship to drug exposure [80]. Denosumab was also proven effective for increasing BMD over 2 years in women receiving adjuvant aromatase inhibitor therapy for breast cancer [81] and for increasing BMD and reducing the incidence of vertebral fractures over 3 years in men with non-metastatic prostate cancer receiving androgen deprivation therapy [82]. The only other RANKL-inhibitor currently under clinical development we are aware of is ALX-0141, a nanobody with Phase I results presented in 2011 [83].
Cathepsin K inhibitors
Odanacatib is the most advanced cathepsin K inhibitor currently under development. Odanacatib was shown to be orally bio-available, highly selective for and reversibly binding to cathepsin K [84]. Cathepsin K selectivity is responsible for the lack of accumulation of undesirable collagen in cutaneous fibroblasts [84]. The development of less selective cathepsin K inhibitors, such as balicatib, was stopped in phase 2 development due to the appearance of sclerodermia-like skin lesions [85]. Based on phase I and II results, the recommended dosage is 50mg once weekly per os [86, 87]. The increases in spine and hip BMD observed with odanacatib were comparable to those observed with the bisphosphonate zoledronate and the RANKL-inhibitor denosumab [5, 18, 87]. Interestingly, while there was a smaller reduction in markers of bone resorption in comparison with other powerful antiresorptive agents, the reduction in levels of formation markers was much smaller [88]. Furthermore, histomorphometry of bone biopsies performed in a subset of 32 patients included in the phase II trial showed that the modest reduction in bone formation markers was not accompanied by a suppression of the bone formation rate [87–89]. These findings suggest a decoupling between bone formation and resorption. It was hypothesised that as the inhibition of cathepsin K suppresses osteoclast function but does not impair osteoclast viability, it may preserve the osteoclast-osteoblast crosstalk and maintain bone formation [89]. In addition, unlike conventional antiresorptives, odanacatib displayed site specific effects on trabecular versus cortical bone formation with marked increases in periosteal bone formation and cortical thickness in ovariectomised monkeys [90]. Although their clinical relevance remains to be confirmed, these findings would represent a major advance in the field of bone research. A randomised, placebo-controlled phase III fracture endpoint trial, which has enrolled more than 16000 postmenopausal women with low bone mass, is currently ongoing with expected results during summer 2012 (NCT00529373). Once available, the results of this study will unveil a comprehensive efficacy and safety profile of odanacatib for the treatment of postmenopausal osteoporosis. Another cathepsin K inhibitor currently in phase II of development is ONO-5334 [91].
Access to therapy – another frontier?
Several new therapeutic classes against osteoporosis, for which there is an important medical need, will become available in the near future. Based on today’s known or expected profile of drugs such as the cathepsin K inhibitor odanacatib and the sclerostin antibodies, which both may for the first time allow for a decoupling between bone formation and resorption, it should be anticipated that our options for successfully treating osteoporosis, that is for reducing fracture risk, will be considerably enhanced. Whether new and even better drugs alone will suffice to successfully address the socioeconomical challenges ahead is questionable. Keeping in mind that Switzerland, as one of the leading countries with regard to the proportion of elderly and very old in its population, represents a paradigm for the near future of many other industrialised countries, the results of a recent survey conducted at the emergency wards of 8 Swiss hospitals are enlightening: among 3667 patients who presented with a fragility fracture, only 24% of the women and 14% of the men underwent appropriate diagnostic workup (i.e., measurement of bone mineral density (BMD) by DXA and were adequately prescribed a treatment with a bone active substance proven to reduce fracture risk [92]. Thus, access to diagnosis and adequate therapy is and will supposedly remain a key challenge in Switzerland as well as in other industrialised countries. The development and calibration of the FRAX® fracture risk calculator to the Swiss setting (available under http://www.shef.ac.uk/FRAX/) represented a critical and important step for simplifying the identification of patients at increased risk of fracture [93, 94]. The FRAX® tool has been developed by World Health Organization to evaluate individual fracture risk. It is based on individual patient models that integrate the risks associated with clinical risk factors as well as bone mineral density (BMD) at the femoral neck. The FRAX® algorithms give the 10-year probabilities of hip fracture and of a major osteoporotic fracture (clinical spine, forearm, hip or shoulder fracture) [76, 95]. Country-specific cost-effective FRAX® intervention thresholds have been established for Switzerland [96] and other industrialised countries [97].
Conclusion
Osteoporosis is a highly prevalent severe disease imposing a heavy burden of suffering on patients and costs to society. Osteoporosis can be diagnosed by DXA and the individual fracture risk can be calculated with FRAX®. FRAX®-based cost-effective intervention thresholds exist and are on their way for implementation in daily practice. Therapies proven to reduce fracture risk, even if imperfect, are already available. More effective and better tolerated therapies will become available soon, which may contribute to increase the currently low treatment rate of even severe osteoporosis by allowing tailor-made approaches aimed at minimising fracture risk at the individual patient level.
Acknowledgements:I'm grateful to Dr. Philippe Kress, Glattbrugg, and Romain Perrelet from our institution for their valuable contribution to the manuscript and graphics.
References
1 Consensus development conference: diagnosis, prophylaxis, and treatment of osteoporosis. Am J Med. 1993;94:646–50.
2 Bundesamt für Statistik: Mortality and Life Expectancy tables. http://www.bfs.admin.ch/bfs/portal/de/index/themen/01/06/blank/key/04.html. Last visited February 20, 2010.
3 Bundesamt für Statistik. Szenarien zur Bevölkerungsentwicklung der Schweiz 2005–2050. http://www.bfs.admin.ch/bfs/portal/de/index/news/publikationen.Document.83713.pdf. Last visited May 14, 2008.
4 Lippuner K, Grifone S, Schwenkglenks M, Schwab P, Popp AW, Senn C, Perrelet R. (2011) Comparative trends in hospitalizations for osteoporotic fractures and other frequent diseases between 2000 and 2008. Osteoporos Int
5 Black DM, Delmas PD, Eastell R, et al. Once-yearly zoledronic acid for treatment of postmenopausal osteoporosis. N Engl J Med. 2007;356:1809–22.
6 Black DM, Thompson DE, Bauer DC, Ensrud K, Musliner T, Hochberg MC, et al. Fracture risk reduction with alendronate in women with osteoporosis: the Fracture Intervention Trial. FIT Research Group. J Clin Endocrinol Metab. 2000;85:4118–24.
7 Cummings SR, Black DM, Thompson DE, et al. Effect of alendronate on risk of fracture in women with low bone density but without vertebral fractures: results from the Fracture Intervention Trial. JAMA. 1998;280:2077–82.
8 McClung MR, Geusens P, Miller PD, et al. Effect of risedronate on the risk of hip fracture in elderly women. Hip Intervention Program Study Group. N Engl J Med. 2001;344:333–40.
9 Harris ST, Watts NB, Genant HK, et al. Effects of risedronate treatment on vertebral and nonvertebral fractures in women with postmenopausal osteoporosis: a randomized controlled trial. Vertebral Efficacy With Risedronate Therapy (VERT) Study Group. JAMA. 1999;282:1344–52.
10 Black DM, Schwartz AV, Ensrud KE, et al. Effects of continuing or stopping alendronate after 5 years of treatment: the Fracture Intervention Trial Long-term Extension (FLEX): a randomized trial. JAMA. 2006;296:2927–38.
11 Rizzoli R, Burlet N, Cahall D, et al. Osteonecrosis of the jaw and bisphosphonate treatment for osteoporosis. Bone. 2008;42:841–7.
12 Rizzoli R, Akesson K, Bouxsein M, Kanis JA, Napoli N, Papapoulos S, et al. Subtrochanteric fractures after long-term treatment with bisphosphonates: a European Society on Clinical and Economic Aspects of Osteoporosis and Osteoarthritis, and International Osteoporosis Foundation Working Group Report. Osteoporos Int. 2011;22:373–90.
13 Shane E, Burr D, Ebeling PR, et al. Atypical subtrochanteric and diaphyseal femoral fractures: report of a task force of the American Society for Bone and Mineral Research. J Bone Miner Res. 2010;25:2267–94.
14 Ettinger B, Black DM, Mitlak BH, et al. Reduction of vertebral fracture risk in postmenopausal women with osteoporosis treated with raloxifene: results from a 3-year randomized clinical trial. Multiple Outcomes of Raloxifene Evaluation (MORE) Investigators. JAMA. 1999;282:637–45.
15 Martino S, Cauley JA, Barrett-Connor E, Powles TJ, Mershon J, Disch D, et al. Continuing outcomes relevant to Evista: breast cancer incidence in postmenopausal osteoporotic women in a randomized trial of raloxifene. J Natl Cancer Inst. 2004;96:1751–61.
16 Barrett-Connor E, Mosca L, Collins P, Geiger MJ, Grady D, Kornitzer M, et al. Effects of raloxifene on cardiovascular events and breast cancer in postmenopausal women. N Engl J Med. 2006;355:125–37.
17 Vogel VG, Costantino JP, Wickerham DL, et al. Effects of tamoxifen vs raloxifene on the risk of developing invasive breast cancer and other disease outcomes: the NSABP Study of Tamoxifen and Raloxifene (STAR) P-2 trial. JAMA. 2006;295:2727–41.
18 Cummings SR, San Martin J, McClung MR, et al. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med. 2009;361:756-65.
19 Terpos E, Dimopoulos MA. Interaction between the skeletal and immune systems in cancer: mechanisms and clinical implications. Cancer immunology, immunotherapy: CII 2011;60:305–17.
20 Toulis KA, Anastasilakis AD. Increased risk of serious infections in women with osteopenia or osteoporosis treated with denosumab. Osteoporos Int. 2010;21:1963–4.
21 Compston J. Pathophysiology of atypical femoral fractures and osteonecrosis of the jaw. Osteoporos Int. 2011;22:2951–61.
22 Papapoulos S, Chapurlat R, Libanati C, et al. Five years of denosumab exposure in women with postmenopausal osteoporosis: Results from the first two years of the FREEDOM extension. J Bone Miner Res 2011.
23 Kyrgidis A, Toulis KA. Denosumab-related osteonecrosis of the jaws. Osteoporos Int. 2011;22:369–70.
24 Saad F, Brown JE, Van Poznak C, et al. (2011) Incidence, risk factors, and outcomes of osteonecrosis of the jaw: integrated analysis from three blinded active-controlled phase III trials in cancer patients with bone metastases. Ann Oncol
25 Baron R, Ferrari S, Russell RG. Denosumab and bisphosphonates: different mechanisms of action and effects. Bone. 2011;48:677–92.
26 Greenspan SL, Bone HG, Ettinger MP, Hanley DA, Lindsay R, Zanchetta JR, et al. Effect of recombinant human parathyroid hormone (1-84) on vertebral fracture and bone mineral density in postmenopausal women with osteoporosis: a randomized trial. Ann Intern Med. 2007;146:326–39.
27 Neer RM, Arnaud CD, Zanchetta JR, et al. Effect of parathyroid hormone (1-34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med. 2001;344:1434–441.
28 Saag KG, Shane E, Boonen S, Marin F, Donley DW, Taylor KA, et al. Teriparatide or alendronate in glucocorticoid-induced osteoporosis. N Engl J Med. 2007;357:2028–39.
29 Manolagas SC. Birth and death of bone cells: basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr Rev. 2000;21:115–37.
30 Jilka RL, Weinstein RS, Bellido T, Roberson P, Parfitt AM, Manolagas SC. Increased bone formation by prevention of osteoblast apoptosis with parathyroid hormone. J Clin Invest. 1999;104:439–46.
31 Brown EM, Gamba G, Riccardi D, Lombardi M, Butters R, Kifor O, et al. Cloning and characterization of an extracellular Ca(2+)-sensing receptor from bovine parathyroid. Nature. 1993;366:575–80.
32 Steddon SJ, Cunningham J. Calcimimetics and calcilytics – fooling the calcium receptor. Lancet. 2005;365:2237–9.
33 Baron R, Rawadi G. Targeting the Wnt/beta-catenin pathway to regulate bone formation in the adult skeleton. Endocrinology. 2007;148:2635–43.
34 Nusse R, Fuerer C, Ching W, Harnish K, Logan C, Zeng A, et al. Wnt signaling and stem cell control. Cold Spring Harbor symposia on quantitative biology 2008;73:59–66.
35 Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781–810.
36 Rey JP, Ellies DL. Wnt modulators in the biotech pipeline. Developmental dynamics: an official publication of the American Association of Anatomists 2010;239:102–14.
37 Noble BS. The osteocyte lineage. Arch Biochem Biophys. 2008;473:106–11.
38 van Bezooijen RL, ten Dijke P, Papapoulos SE, Lowik CW. SOST/sclerostin, an osteocyte-derived negative regulator of bone formation. Cytokine Growth Factor Rev. 2005;16:319–27.
39 Martin RB. Toward a unifying theory of bone remodeling. Bone. 2000;26:1–6.
40 Robling AG, Niziolek PJ, Baldridge LA, et al. Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J Biol Chem. 2008;283:5866–75.
41 Lin C, Jiang X, Dai Z, Guo X, Weng T, Wang J, et al. Sclerostin mediates bone response to mechanical unloading through antagonizing Wnt/beta-catenin signaling. J Bone Miner Res. 2009;24:1651–61.
42 Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423:337–42.
43 McClung MR. Inhibition of RANKL as a treatment for osteoporosis: preclinical and early clinical studies. Curr Osteoporos Rep. 2006;4:28–33.
44 Vaananen HK, Laitala-Leinonen T. Osteoclast lineage and function. Arch Biochem Biophys. 2008;473:132–8.
45 Bromme D, Lecaille F. Cathepsin K inhibitors for osteoporosis and potential off-target effects. Expert Opin Investig Drugs. 2009;18:585–600.
46 Gelb BD, Shi GP, Chapman HA, Desnick RJ. Pycnodysostosis, a lysosomal disease caused by cathepsin K deficiency. Science. 1996;273:1236–8.
47 Saftig P, Hunziker E, Wehmeyer O, Jones S, Boyde A, Rommerskirch W, et al. Impaired osteoclastic bone resorption leads to osteopetrosis in cathepsin-K-deficient mice. Proc Natl Acad Sci U S A 1998;95:13453–8.
48 Rizzoli R, Kraenzlin M, Krieg MA, Mellinghoff HU, Lamy O, Lippuner K. Indications to teriparatide treatment in patients with osteoporosis. Swiss Med Wkly. 2011;141:w13297.
49 Daddona PE, Matriano JA, Mandema J, Maa YF. Parathyroid hormone (1-34)-coated microneedle patch system: clinical pharmacokinetics and pharmacodynamics for treatment of osteoporosis. Pharmaceutical research. 2011;28:159–65.
50 Cosman F, Lane NE, Bolognese MA, Zanchetta JR, Garcia-Hernandez PA, Sees K, et al. Effect of transdermal teriparatide administration on bone mineral density in postmenopausal women. J Clin Endocrinol Metab. 2010;95:151–8.
51 Lippuner K, Zehnder HJ, Casez JP, Takkinen R, Jaeger P. PTH-related protein is released into the mother’s bloodstream during location: evidence for beneficial effects on maternal calcium-phosphate metabolism. J Bone Miner Res. 1996;11:1394–9.
52 Casez J, Pfammatter R, Nguyen Q, Lippuner K, Jaeger P. Diagnostic approach to hypercalcemia: relevance of parathyroid hormone and parathyroid hormone-related protein measurements. Eur J Intern Med. 2001;12:344–9.
53 Rosenblatt M. When two keys fit one lock, surprises follow. Nat Chem Biol. 2009;5:707–8.
54 Martin TJ. Osteoblast-derived PTHrP is a physiological regulator of bone formation. J Clin Invest. 2005;115:2322–4.
55 Kimura S, Nakagawa T, Matsuo Y, Ishida Y, Okamoto Y, Hayashi M. JTT-305, an orally active calcium-sensing receptor antagonist, stimulates transient parathyroid hormone release and bone formation in ovariectomized rats. Eur J Pharmacol. 2011;668:331–6.
56 Kumar S, Matheny CJ, Hoffman SJ, et al. An orally active calcium-sensing receptor antagonist that transiently increases plasma concentrations of PTH and stimulates bone formation. Bone. 2010;46:534–42.
57 Brunkow ME, Gardner JC, Van Ness J, et al. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am J Hum Genet. 2001;68:577–89.
58 Balemans W, Ebeling M, Patel N, et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum Mol Genet. 2001;10:537–43.
59 Papapoulos SE. Targeting sclerostin as potential treatment of osteoporosis. Ann Rheum Dis. 2011;70(Suppl 1):i119–22.
60 Beighton P, Durr L, Hamersma H. The clinical features of sclerosteosis. A review of the manifestations in twenty-five affected individuals. Ann Intern Med. 1976;84:393–7.
61 Hamersma H, Gardner J, Beighton P. The natural history of sclerosteosis. Clinical genetics. 2003;63:192–7.
62 Moester MJ, Papapoulos SE, Lowik CW, van Bezooijen RL. Sclerostin: current knowledge and future perspectives. Calcif Tissue Int. 2010;87:99–107.
63 Gardner JC, van Bezooijen RL, Mervis B, Hamdy NA, Lowik CW, Hamersma H, et al. Bone mineral density in sclerosteosis; affected individuals and gene carriers. J Clin Endocrinol Metab. 2005;90:6392–5.
64 Vanhoenacker FM, Balemans W, Tan GJ, Dikkers FG, De Schepper AM, Mathysen DG, et al. Van Buchem disease: lifetime evolution of radioclinical features. Skeletal Radiol. 2003;32:708–18.
65 Van Hul W, Balemans W, Van Hul E, Dikkers FG, Obee H, Stokroos RJ, et al. Van Buchem disease (hyperostosis corticalis generalisata) maps to chromosome 17q12-q21. Am J Hum Genet. 1998;62:391–9.
66 Jacobs P. Van Buchem disease. Postgrad Med J. 1977;53:497–506.
67 Ominsky MS, Vlasseros F, Jolette J, et al. Two doses of sclerostin antibody in cynomolgus monkeys increases bone formation, bone mineral density, and bone strength. J Bone Miner Res. 2010;25:948–59.
68 Li X, Warmington KS, Niu QT, et al. Inhibition of sclerostin by monoclonal antibody increases bone formation, bone mass, and bone strength in aged male rats. J Bone Miner Res. 2010;25:2647–56.
69 Padhi D, Jang G, Stouch B, Fang L, Posvar E. Single-dose, placebo-controlled, randomized study of AMG 785, a sclerostin monoclonal antibody. J Bone Miner Res. 2011;26:19–26.
70 Tian E, Zhan F, Walker R, Rasmussen E, Ma Y, Barlogie B, et al. The role of the Wnt-signaling antagonist DKK1 in the development of osteolytic lesions in multiple myeloma. N Engl J Med. 2003;349:2483–94.
71 Butler JS, Murray DW, Hurson CJ, O’Brien J, Doran PP, O’Byrne JM. The role of Dkk1 in bone mass regulation: correlating serum Dkk1 expression with bone mineral density. J Orthop Res. 2011;29:414–8.
72 Anastasilakis AD, Polyzos SA, Avramidis A, Toulis KA, Papatheodorou A, Terpos E. The effect of teriparatide on serum Dickkopf-1 levels in postmenopausal women with established osteoporosis. Clin Endocrinol. (Oxf) 2010;72:752–7.
73 Gatti D, Viapiana O, Idolazzi L, Fracassi E, Rossini M, Adami S. The waning of teriparatide effect on bone formation markers in postmenopausal osteoporosis is associated with increasing serum levels of DKK1. J Clin Endocrinol Metab. 2011;96:1555–9.
74 (2010) National Osteoporosis Foundation. Clinician’s Guide to Prevention and Treatment of Osteoporosis. http://www.nof.org/professionals/clinical-guidelines.
75 Compston J, Cooper A, Cooper C, Francis R, Kanis JA, Marsh D, et al. Guidelines for the diagnosis and management of osteoporosis in postmenopausal women and men from the age of 50 years in the UK. Maturitas. 2009;62:105–8.
76 Kanis JA, Burlet N, Cooper C, Delmas PD, Reginster JY, Borgstrom F, et al. European guidance for the diagnosis and management of osteoporosis in postmenopausal women. Osteoporos Int. 2008;19:399–428.
77 Enders GH. Wnt therapy for bone loss: golden goose or Trojan horse? J Clin Invest. 2009;119:758–60.
78 Kansara M, Tsang M, Kodjabachian L, et al. Wnt inhibitory factor 1 is epigenetically silenced in human osteosarcoma, and targeted disruption accelerates osteosarcomagenesis in mice. J Clin Invest. 2009;119:837–51.
79 Boonen S, Adachi JD, Man Z, et al. Treatment with denosumab reduces the incidence of new vertebral and hip fractures in postmenopausal women at high risk. J Clin Endocrinol Metab. 2011;96:1727–36.
80 (2011) Prescribing information of Prolia® (denosumab) approved by the FDA. Last revision 09/2011. Available under http://www.accessdata.fda.gov/drugsatfda_docs/label/2011/125320s5s6lbl.pdf. Last accessed February 22, 2012.
81 Ellis GK, Bone HG, Chlebowski R, Paul D, Spadafora S, Smith J, et al. Randomized Trial of Denosumab in Patients Receiving Adjuvant Aromatase Inhibitors for Nonmetastatic Breast Cancer. J Clin Oncol (2008).
82 Smith MR, Egerdie B, Hernandez Toriz N, et al. Denosumab in men receiving androgen-deprivation therapy for prostate cancer. N Engl J Med. 2009;361:745–55.
83 (2011) Safety, pharmacokinetics and efficacy of anti-RANKL Nanobody®ALX-0141 in healthy postmenopausal women. Presentation at the EULAR 2011. Available under http://www.ablynx.com/wp-content/uploads/2011/05/2011-05-27-ALX-0141-EULAR_final.pdf. Last accessed February 22, 2012.
84 Gauthier JY, Chauret N, Cromlish W, et al. The discovery of odanacatib (MK-0822), a selective inhibitor of cathepsin K. Bioorganic & medicinal chemistry letters. 2008;18:923–8.
85 Runger TM, Adami S, Benhamou CL, Czerwinski E, Farrerons J, Kendler DL, et al. Morphea-like skin reactions in patients treated with the cathepsin K inhibitor balicatib. J Am Acad Dermatol. 2012;66:e89–96.
86 Stoch SA, Zajic S, Stone J, et al. Effect of the cathepsin K inhibitor odanacatib on bone resorption biomarkers in healthy postmenopausal women: two double-blind, randomized, placebo-controlled phase I studies. Clin Pharmacol Ther. 2009;86:175–82.
87 Bone HG, McClung MR, Roux C, Recker RR, Eisman JA, Verbruggen N, et al. Odanacatib, a cathepsin-K inhibitor for osteoporosis: a two-year study in postmenopausal women with low bone density. J Bone Miner Res. 2010;25:937–47.
88 Perez-Castrillon JL, Pinacho F, De Luis D, Lopez-Menendez M, Duenas Laita A. (2010) Odanacatib, a new drug for the treatment of osteoporosis: review of the results in postmenopausal women. Journal of osteoporosis 2010:
89 Rachner TD, Khosla S, Hofbauer LC. Osteoporosis: now and the future. Lancet. 2011;377:1276–87.
90 Cusick T, Chen CM, Pennypacker BL, Pickarski M, Kimmel D, Scott BB, et al. (2011) Odanacatib treatment increases hip bone mass and cortical thickness by preserving endocortical bone formation and stimulating periosteal bone formation in the ovariectomized adult rhesus monkey. J Bone Miner Res
91 Eastell R, Nagase S, Ohyama M, Small M, Sawyer J, Boonen S, et al. Safety and efficacy of the cathepsin K inhibitor ONO-5334 in postmenopausal osteoporosis: the OCEAN study. J Bone Miner Res. 2011;26:1303–12.
92 Suhm N, Lamy O, Lippuner K. Management of fragility fractures in Switzerland: results of a nationwide survey. Swiss Med Wkly. 2008;138:674–83.
93 Lippuner K, Johansson H, Kanis JA, Rizzoli R. Remaining lifetime and absolute 10-year probabilities of osteoporotic fracture in Swiss men and women. Osteoporos Int. 2009;20:1131–40.
94 Lippuner K, Johansson H, Kanis JA, Rizzoli R. FRAX assessment of osteoporotic fracture probability in Switzerland. Osteoporos Int. 2010;21:381–9.
95 Kanis JA, Hans D, Cooper C, et al. Interpretation and use of FRAX in clinical practice. Osteoporos Int. 2011;22:2395–411.
96 Lippuner K, Johansson H, Borgstrom F, Kanis JA, Rizzoli R. (2012) Cost-effective intervention thresholds against osteoporotic fractures based on FRAX(R) in Switzerland. Osteoporos Int
97 Tosteson AN, Melton LJ, 3rd, Dawson-Hughes B, Baim S, Favus MJ, Khosla S, et al. Cost-effective osteoporosis treatment thresholds: the United States perspective. Osteoporos Int. 2008;19:437–47.