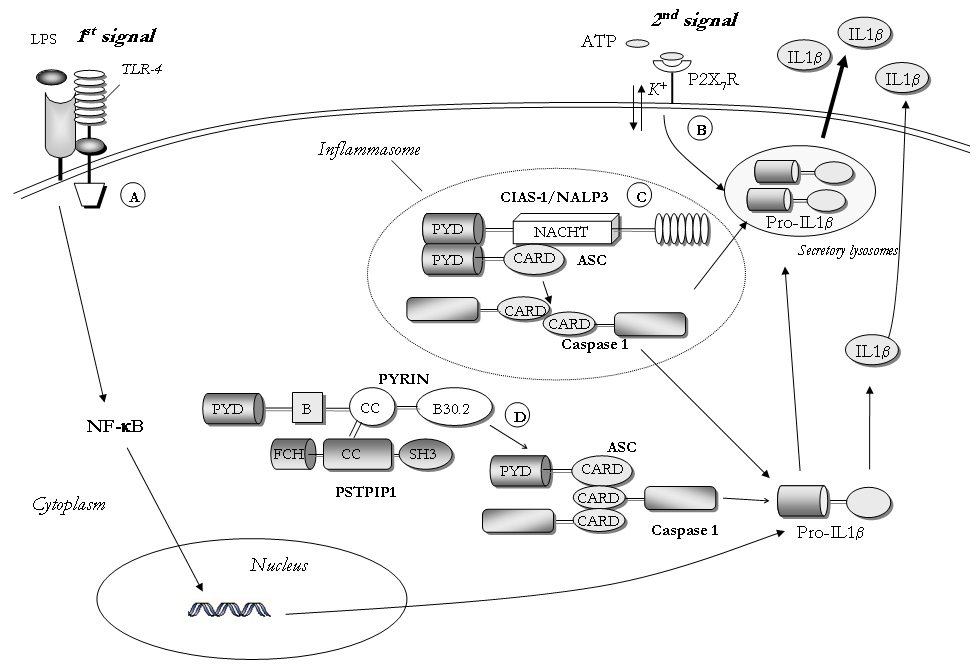
Figure 1
Role of NOD2/CARD15 in the activation of NF-κB and caspase 1.
DOI: https://doi.org/10.4414/smw.2012.13602
Under the term monogenic autoinflammatory syndromes are subsumed a number of different conditions secondary to mutations of genes coding for proteins that play a pivotal role in the regulation of the inflammatory response.
Due to their genetic nature, most of these disorders have an early onset, ranging from the first hours to the second decade of life.
Only a limited number of patients experience a disease onset during adulthood.
Even if nowadays there is much more awareness of these disorders, the extreme rarity and relatively recent identification as autonomous entities, often result in delayed diagnosis.
Clinically the autoinflammatory syndromes are characterised by recurrent flares of systemic inflammation presenting in the majority of cases as sudden fever episodes associated with elevation of acute phase reactants together with a number of clinical manifestations such as rash, serositis (peritonitis, pleurisy), lymphadenopathy and arthritis.
Symptom-free intervals are characterised by complete wellbeing, normal growth and complete normalisation of acute phase reactants.
Familial Mediterranean fever (FMF, MIM 249100), mevalonate-kinase deficiency (MKD, MIM 260920) and tumour necrosis factor (TNF) receptor-associated periodic syndrome (TRAPS, MIM 142680) are the three monogenic disorders subsumed under the term periodic fevers. A systemic inflammation dominated by a characteristic urticarial rash associated with a number of other clinical symptoms is, on the other hand, typical of familial cold autoinflammatory syndrome (FCAS, MIM 120100), Muckle-Wells syndrome (MWS, MIM 191900) and chronic infantile neurological cutaneous and articular syndrome (CINCA, MIM 607115). These diseases represent the clinical spectrum of different mutations of a gene named cold-induced autoinflammatory syndrome 1 (CIAS-1, or NLRP3) coding for a protein called cryopyrin. Hence these disorders are also known under the term cryopyrin-associated periodic syndromes (CAPS).
Other conditions are characterised by the appearance of typical granulomatous formations (granulomatous disorders). Blau’s syndrome (MIM 186580), also known as familial juvenile systemic granulomatosis, presents with non-caseating granulomatous inflammation affecting the joint, skin, and uveal tract (the triad of arthritis, dermatitis and uveitis) and is associated with mutations of the NACHT domain of the gene CARD15 (or NOD2). Of note is that mutations in this same gene have been associated with Crohn’s disease, another granulomatous disease.
Finally, other rare disorders dominated by the presence of sterile pyogen abscesses chiefly affecting skin, joints and bones (pyogenic disorders) include the PAPA syndrome (pyogenic sterile arthritis, pyoderma gangrenosum and acne) (MIM 604416), associated with mutations of the CD2-binding protein 1 (CD2BP1 or PSTPIP1) gene, the Majeed syndrome (MIM 609628), characterised by chronic recurrent multifocal osteomyelitis, congenital dyserythropoietic anaemia and neutrophilic dermatosis, caused by mutations of LPIN2 gene, and deficiency of the interleukin-1receptor antagonist (DIRA) (MIM 612852), a recently identified autosomal recessive autoinflammatory syndrome characterised by severe systemic inflammation beginning approximately at the time of birth with multifocal osteomyelitis, periostitis and pustulosis and caused by mutations of the IL1RN gene encoding interleukin-1 receptor antagonist.
Familial Mediterranean fever was first described as a distinct entity in the second part of the twentieth century [1].
Figure 1
Role of NOD2/CARD15 in the activation of NF-κB and caspase 1.
It is the most frequent among hereditary recurrent inflammatory disorders.
It affects populations of Mediterranean descent: Arabs from the East as well as from the West, Armenians, Turks, non-Ashkenazi and other Jews, Druzes, Lebanese, Italians and Greeks.
Among non-Ashkenazi Jews and Turks, the frequency of heterozygotes in the general population is very high, ranging from 1:3 to 1:6 [2].
In 1992, the gene associated with FMF was mapped to the short arm of chromosome 16 and cloned in 1997 [3, 4]. It was called MEFV for MEditerranean FeVer.
The full-length transcript of 3.7 Kb encodes a protein called pyrin/marenostrin consisting of 781 amino acids, that has been suggested as playing a role in the regulation of the so-called inflammasome (fig. 1). The inflammasome is a cytoplasmatic multiprotein complex that plays a crucial part in the production and secretion of pro-inflammatory cytokines, such as interleukin (IL)-1β [5], and is involved in the pathogenesis of various autoinflammatory diseases [6] (see below).
Several domains of the protein are remarkable: the pyrin domain is a specific domain of 90 amino acids located in the N-terminal region, and defines a novel class of proteins called the pyrin family. A second domain called B30.2 or SPRY is located in the C-terminal region of the protein and contains the most frequent mutations associated with FMF.
Some pathogenetic data relating to the function of these domains have been established.
First, it has been shown that pyrin can interact with ASC (apoptotic speck protein) by homotypic pyrin binding domain; ASC mediates both NF-κB and pro-caspase-1 activation with associated processing and secretion of interleukin IL-1β, and apoptosis [7].
The second point derives from the discovery of the inflammasome.
Early works suggest that pyrin modulates the inflammasome by interacting both with its pyrin and B30.2 domains (fig. 1). Recently it has also been found that PSTPIP1, the protein involved in PAPA syndrome (see below), can activate pyrin, leading to the interaction between PYD and ASC, ASC dimerisation and recruitment and activation of caspase-1, independently of activation of the NLRP3-inflammasome (fig. 1). This hypothesis has recently been confirmed in FMF animal models [8].
Almost ⅔ of patients have a disease onset before 5 years and almost all patients before the second decade of life [9–11].
In FMF fever attacks last from a few hours to 3 or 4 days, and are typically associated with signs of acute serosal inflammation. Peritonitis and monolateral pleurisy are present in more than 90 and 40% of patients respectively and are the cause of two of the most typical symptoms of FMF: severe abdominal pain and chest pain. Scrotitis and pericarditis may also be present in a minority of patients.
Large joints may be affected by arthritis or arthralgia in more than 50% of patients (chiefly knees, hips and ankles), and the erysipelas-like erythema of the lower limbs, which represents the most common skin lesion, is present in almost 25% of cases.
Attacks resolve spontaneously, and there is no regular periodicity of recurrences.
Their frequency varies considerably from one patient to another and, in the same patient, from one period of life to another.
Some factors may act as triggers of the inflammatory attacks, especially stress, viral disease or even drugs such as metaraminol and cisplatin. Prodromes of FMF attacks may include discomfort at the impending attack site or various constitutional, emotional, and physical complaints, including irritability, dizziness, increased appetite, and altered taste sensation. A prodrome is a valid sign of an imminent attack in a subgroup of patients with FMF.
Except amyloidosis, which represents the major complication of the disease, chronic manifestations such as encapsulating peritonitis and chronic destructive arthritis affecting hips and knees in particular are rare. Splenomegaly, usually without specific consequences, may also be observed in a subgroup of patients with incompletely controlled inflammation.
Even if genetic testing is available, the diagnosis of FMF relies mainly on clinical arguments. Different sets of criteria have been developed in countries presenting a high prevalence of the disease [12, 13] but they have not yet been validated in populations presenting other autoinflammatory syndromes.
Molecular analysis oftheMEFV gene provides genetic confirmation of the diagnosis, but it should be noted that interpretation of the molecular analysis can sometimes be difficult.
In the clinical context of FMF, the presence of 2 mutations on different alleles (homozygosity or compound heterozygosity) makes it possible to confirm the diagnosis but, at the same time, it cannot be ruled out in a patient with a clinical presentation consistent with the disease even if carrying one MEFVmutation only.
Although five mutations represent more than 85% of all the mutations, some rare or unknown mutations exist (http://fmf.igh.cnrs.fr/infevers/).
Before the colchicine era amyloid nephropathy was the main cause of death in FMF patients.
FMF-associated amyloidosis is a typical form of inflammatory (or AA) amyloidosis. It generally occurs in patients with severe inflammatory attacks beginning early in life (FMF phenotype 1) but may rarely occur even in patients with no recognised clinical inflammatory crisis [14] (FMF phenotype 2).
Increased SAA and C reactive protein between the FMF attacks represent a risk factor for the future development of AA amyloidosis [14, 15] .
It has been established that the prevalence of amyloidosis varies according to ethnic groups, suggesting that genetic (presence of M694V mutation in the homozygous state, SAA polymorphisms) and/or environmental factors participate in the occurrence of amyloidosis in the disease [16, 17].
Daily colchicine is the treatment of choice to prevent recurrence of attacks and occurrence of amyloidosis [18, 19]. The usual dose of colchicine is 1 mg/day. If the disease activity is not controlled, due either to recurrence of attacks or persistent elevated inflammatory parameters, especially SAA, the colchicine dose should be increased by 0.5 mg per day every 3–6 months up to 2.5 mg per day.
Diarrhoea due to colchicine is rare and can be managed by dividing the daily dose in two. A recent study suggests that in children the optimal colchicine dosage, i.e. that which reduces the frequency of attacks, ESR, CRP and fibrinogen levels during the attack-free periods can be calculated on the basis of body weight and body surface area [20]. In some cases, proteinuria due to amyloidosis disappears under treatment with colchicine [21]. Although colchicine intoxication remains a severe threat, long term daily colchicine is a relatively safe treatment. Adverse effects of colchicine on sperm function are controversial and long term use of colchicine can be considered as globally safe including during pregnancy.
True non-responders to colchicine are very rare; the majority are non-compliant patients.
In these non-responders, no treatment has proven its efficacy. Interferon alpha has been proposed, but promising early results have not been confirmed. More recently, anti-IL-1 blockers, such as anakinra and canakinumab have shown efficacy in some patients who were resistant to colchicine, in agreement with the pathogenic data obtained on the role of pyrin in the IL-1 secretion pathway. The use of IL-1 blockers is however not validated and further studies are needed to determine the actual indication for their employment in FMF and their safety in the long run. TNF inhibitors have also been tried with success in some patients.
Periodic fever associated with mevalonate kinase deficiency (MKD) was originally identified in 1984 in six patients of Dutch ancestry with a long history of recurrent attacks of fever of unknown cause and a high serum IgD level [22]. For this reason this disorder has also been named hyper IgD syndrome or Dutch fever.
High IgD plasma levels were considered to be a diagnostic hallmark until mutations in the mevalonate kinase (MVK) gene, encoded on chromosome 12q24, were identified as the cause of the disease [23].
The complete deficiency of this enzyme causes a distinct syndrome called mevalonic aciduria (MA; MIM 251170), which is clinically characterised by severe mental retardation, ataxia, failure to thrive, myopathy and cataracts; notably, these patients also suffer from recurrent fever attacks. MVK is an essential enzyme in the isoprenoid biosynthesis pathway which produces several biomolecules involved in different cellular processes.
After identification of the molecular defect it became clear that the distribution of MKD is not limited to northern European populations. A relevant number of patients have also been observed among populations living around the Mediterranean basin [24] and Asia [25]. Moreover, due to the low sensitivity and specificity of IgD serum levels, the term hyper IgD syndrome has been replaced by periodic fever associated with mevalonate kinase deficiency.
Although the dysregulation of this biochemical pathway seems to play a pivotal role in the development of fever, at present the pathogenetic mechanisms leading to the autoinflammatory disease remain poorly understood. MVK catalyses the ATP-dependent phosphorylation of mevalonate to 5-phosphomevalonate, and is the first enzyme to follow the highly regulated enzyme hydroxymethylglutaryl-coenzyme A (HMG-CoA) reductase in isoprenoid biosynthesis.
Cells from patients with the MKD phenotype still show residual MVK enzyme activities [26, 27] (from 1 to 8% of the activities with respect to control cells), while cells from patients with the MA phenotype show enzyme activity that is below laboratory detection level [28] (approximately 0.1% of normal individuals). This enzymatic deficiency is reflected in the high levels of mevalonic acid in plasma and urine of patients with MA and in the low to moderate levels of mevalonate acid in patients with periodic fever associated with MKD.
The precise molecular mechanism by which the depressed activity of MKV leads to inflammatory flares and fever episodes is still unknown. Two main hypotheses have been formulated: the first suggests that inflammation is related to elevated mevalonic acid levels. This concept was derived from the clinical observation that a reduction in the severity and frequency of fever episodes occurred in 6 MKD adult patients treated with simvastatin. The latter is a competitive inhibitor of HMG-CoA reductase which would result in lowering of mevalonate levels and a consequent reduction in inflammation.
In contrast, other in vitro studies have shown that shortage of isoprenoid end products, rather than an excess of mevalonate, contributes to inflammatory responses in MKD. Consistent with this theory, the shortage of non-sterol isoprenoid end products, mainly the geranylgeranyl groups, led to increased activation of caspase 1 in circulating monocytes with consequent hypersecretion of the 17 Kd active form of IL-1β [29].
MKD is essentially a paediatric disease. Its onset is very early in life, most often in infancy, and almost all patients develop the disease within the first decade of life.
Fever attacks have an abrupt onset and last 4–6 days. Severe abdominal pain often accompanied by vomiting and/or diarrhoea is the most frequent manifestation associated with fever attacks.
Irritability and cervical lymphadenopathy are common features too and splenomegaly may be found in about half of patients during acute episodes.
Axillary, inguinal and intra-abdominal lymph node enlargement may also be present. Mucocutaneous manifestations are frequent and include erythematous macules, urticaria-like lesions and, less commonly, oral aphthous lesions. Articular involvement occurs in the majority of patients as arthralgia or as oligoarticular, usually symmetrical, arthritis [24, 30, 31].
The symptoms of MKD persist for years but usually tend to become less pronounced with time. However, in some patients the disease may persist until adulthood.
Although amyloidosis was not considered to be a possible long-term complication of MKD(30), it has recently been described in a few patients [24, 32].
Neutrophilia and elevated acute phase reactants are present during fever attacks.
Increased plasma levels of IgD (>100 UI/ml) during fever episodes and in basal conditions have been considered in the past as a hallmark of the disease. However, the specificity of this finding is low [24, 33].
Concomitant elevation of IgA has been also reported [24, 34].
Increased urinary excretion of mevalonic acid is observed during fever spikes, and decreased MVK activity may also be a pointer to the diagnosis. However, the aforementioned determinations need highly specialised laboratory work and, therefore, are often difficult to perform as screening tests.
Thus the decision to perform the molecular analysis of MVK gene in a child with periodic fever is usually taken on clinical grounds.
MKD is an autosomal recessive disease. So far more than 130 substitutions or deletions of the MVK gene have been reported [35] (http://fmf.igh.cnrs.fr/infevers/).
Some variants (i.e., V310M, A334T) are closely associated with a severe MA phenotype and severely impaired cellular MVK activity [36].
The most common mutation in MVK gene is the V377I variant, which is exclusively associated with the mild phenotype of MKD with some residual MVK activity [26]. It is found in a compound heterozygous state in the vast majority of patients with MKD [24, 36]. Other mutations such as H20P and I268T have also been associated either with MA and MKD phenotype (http://fmf.igh.cnrs.fr/infevers/). It is, therefore, conceivable that some patients may also present an intermediate phenotype, characterised by the typical fever attacks associated with some neurological manifestations (mental retardation, cerebellar ataxia) of variable severity [37]. It is therefore clear that mevalonic aciduria and periodic fever associated with MKD represent the two extremities of a wide clinical spectrum.
Fever attacks usually respond dramatically to the administration of steroids (prednisone: 1 mg/kg/day in a single dose or with a short course of 3 to 5 days). However, due to the high frequency of the fever episodes, some patients may need almost continuous treatment. Thalidomide has proven to be ineffective [38].
The use of biological treatments is largely anecdotal and sometimes controversial.
Anti-TNF therapy has been found to reduce the frequency and intensity of fever attacks in some patients [39] but not in others [40] Recently, the use of the IL-1 receptor antagonist (anakinra) was found to be effective in a patient [41]. However, these observations require confirmation in large multicentre therapeutic trials.
A dominant mode of inheritance and long-lasting fever episodes identify an other periodic fever syndrome, defined by the acronym TRAPS, caused by mutations in the p55 TNF receptor (or TNFR1A), encoded by the TNF super family receptor 1A gene [42] (TNFRSF1A). Although TRAPS was initially described in subjects of Nordic origin [43], as emphasised by the name familial Hibernian fever, mutations in TNFRSF1Ahave been found in many populations, including Black Americans, Japanese and those of Mediterranean ancestry, where FMF is highly prevalent.
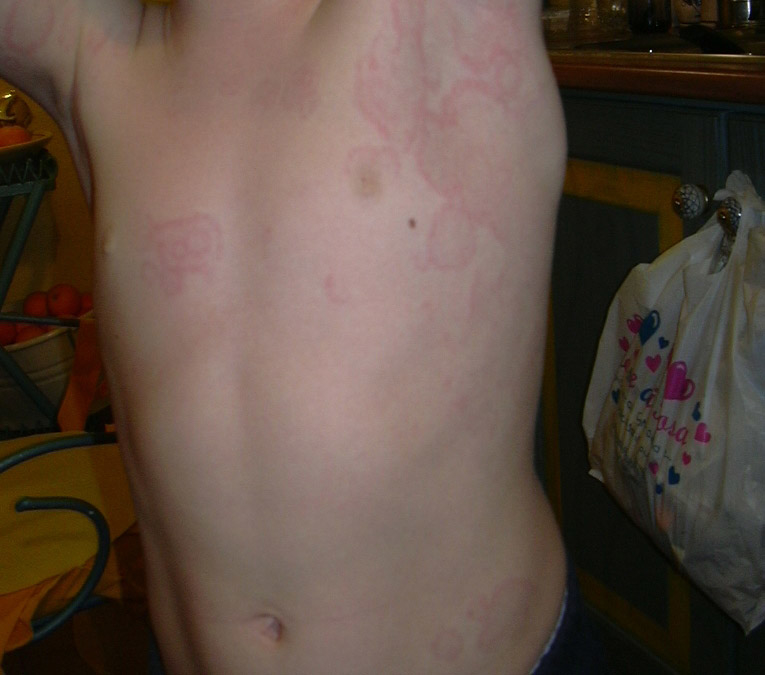
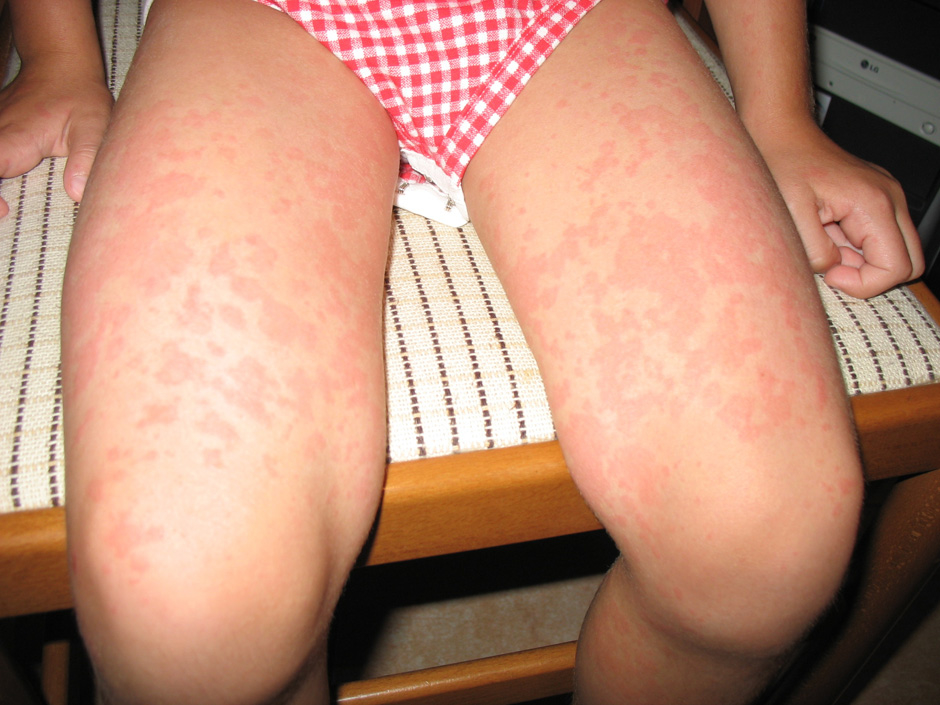
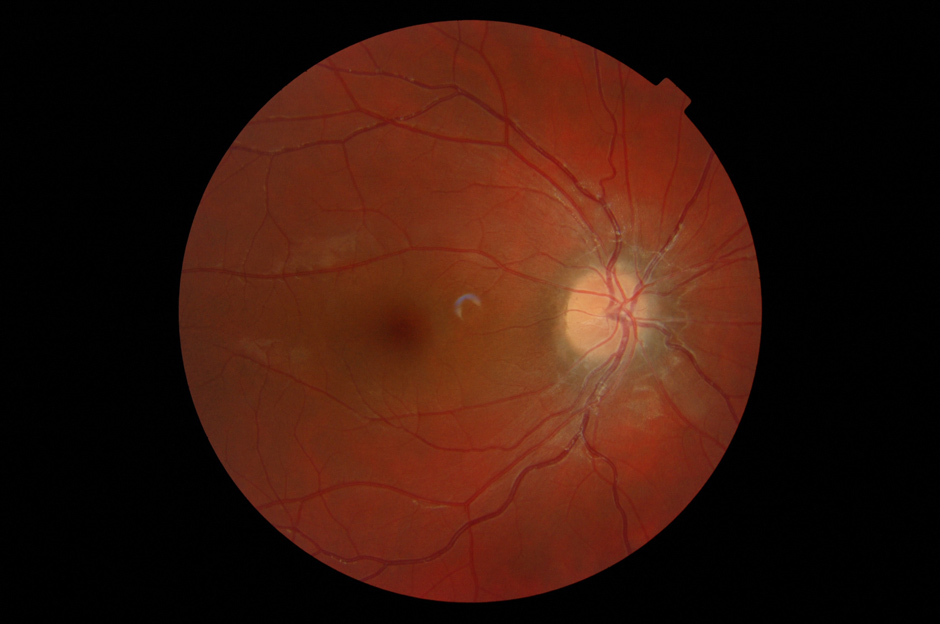
Figure 2
(a) Skin rash in a TRAPS patient; (b) Typical urticarial rash in a Muckle Wells patient carryng the D303N mutation; (c) Chronic papilloedema in a CINCA patient; (d) Typical boggy synovitis in a patient affected with Blau’s disease; (e) Bilateral pyogenic sterile arthritis of the knees in a 4-year-old patient with PAPA syndrome.
A total of 114 sequence variants of the TNFRSF1A have been recorded so far; of which 75 are associated with a TRAPS phenotype [35] (http://fmf.igh.cnrs.fr/infevers/)
The majority of TRAPS-related mutations are missense mutations resulting in single amino acid substitutions in the cysteine rich domains (CRD), CRD1, CRD2, or CRD3 of the ectodomain of the mature TNFR1 (also called p55 TNFR) protein [35, 44, 45].
These CRDs are involved in disulphide bond formation and in the folding of the extracellular portion of the protein. Hence mutations resulting in cysteine substitutions demonstrate a higher penetrance, usually being associated with a more aggressive phenotype and increased probability of developing renal amyloidosis compared to mutations not involving the CRD.
The status of two sequences, R92Q and P46L, has not been fully determined. P46L appears rather as a benign polymorphism and R92Q behaves in the main as an incomplete penetrance mutation [45–47].
In some patients, plasma concentrations of the soluble form of the receptor are low or paradoxically normal during attacks, and may also be low in between. This suggests a quantitative or qualitative abnormality of the soluble form of the receptor. Actually, the shedding of free TNFRs from the membrane produces a pool of soluble receptors which may scavenge circulating TNF by competing with membrane bound receptors. This latter phenomenon represents an important strategy for regulation of the effect of circulating free TNF during acute inflammation. It has been suggested that someTNFRSF1A mutations may interfere with the process of shedding [42], leading to a lack of appropriate TNF inhibition and therefore to uncontrolled inflammation. However, a defect of shedding of the receptor is not observed in all the mutations, suggesting that additional mechanisms could be involved in the pathogenesis of the disease.
At variance with the p75 receptor, p55 TNFR is also able to induce cell apoptosis, via activation of the caspase cascade. In fact, TNFR1 can trigger cellular activation via NF-κB or apoptosis via activation of pro-apoptotic caspases. A defect of TNF-induced apoptosis has been identified in TRAPS patients [46, 48]. This defect observed in patients carrying structural TNFRSF1A mutations may represent an additional mechanism explaining the sustained activation of inflammatory cells during fever episodes.
Through transfection of the mutant form of TNFRI protein in different cell types it was possible to identify an additional relevant pathogenic mechanism related to the diseases. The mutated TNFRI does indeed display a defect of trafficking to the cell membrane with a clear accumulation in the endoplasmic reticulum. Currently the possible pathogenic consequences of ER retention of mutated TNFR1 are under intensive investigation. Increased activation of pro-inflammatory MAP kinases secondary to a stress-induced overproduction of mitochondrial reactive oxygen species (ROS) has recently been shown [49].
TRAPS attacks last generally more than five days and up to three weeks, even though attacks shorter than 5 days have been reported. Abdominal pain can simulate a surgical event; a wide spectrum of skin rashes can be observed in most patients: urticaria-like, plaques and patches (fig. 2a).
The most distinctive lesion is an erythematous, swollen, warm and tender plaque of varying size with hazy edges. It tends to involve the upper and lower limbs but can be observed at the chest. Usually, the rash has a migratory course from the root to the extremity of the limbs.
This pseudo-cellulitis is often accompanied by painful myalgias that represent the other most distinctive manifestation of TRAPS attacks. Thoracic pain, scrotal pain, arthritis, orbital oedema and conjunctivitis are also observed during attacks.
At least in the Caucasian populations, the R92Q mutation is the most frequently observed variant of the TNFRSF1A gene. The majority of children with periodic fever with R92Q mutation display a milder disease course compared to patients carrying structural mutations, with a higher rate of spontaneous resolution or amelioration and much lower prevalence of amyloidosis. Notably, according to different studies, the allele frequency of R92Q variant in the general population ranges from 1.2 to 4%. These clinical observations support the limited pathogenic role of this variant. Based on these data and given the prevalence of this variant in the normal population, great caution should be exercised in interpreting a positive molecular analysis for the R92Q variant, especially in children with periodic fever [50].
When given at the onset of an attack, corticosteroids can attenuate its length and severity. In the most severe forms of the disease, clinical signs of inflammation are almost permanent and require daily use of corticosteroids, leading to dependence and requiring the use of other anti-inflammatory drugs. Colchicine does not seem to prevent recurrences of TRAPS attacks.
TNF inhibitors seem designed as treatment of TRAPS. Etanercept, a TNFRSF1B receptor-immunoglobulin fusion molecule, mimics the effect of the normal soluble TNF receptor and thus compensates its deficit in TRAPS patients. Etanercept and other TNF inhibitors have provided various degrees of clinical improvement and allowed savings of steroids in many cases [51–53]. However, most of the TRAPS patients experience a partial response when treated with Etanercept [54–58].
In other cases the efficacy tends to decrease with time. Interestingly, a paradoxal reaction with exacerbation of the inflammatory signs has been observed after administration of anti-TNF monoclonal antibody (infliximab) in some TRAPS patients and thus this drug should not be used in this indication [59]. Recently it has become clearer that the use of the anti-IL-1 inhibitors (such as recombinant IL-1 receptor antagonist anakinra) can have a better and longer-lasting effect on control of the clinical manifestations [55, 60].
Familial cold autoinflammatory syndrome (FCAS), Muckle-Wells syndrome (MWS) and chronic infantile neurological cutaneous and articular syndrome (CINCA) represent autosomal dominant disorders [61–63] caused by different mutations in a single gene, NLRP3 (NOD-like receptor 3, also known as cold-induced autoinflammatory syndrome 1, CIAS1), encoding a protein called cryopyrin [65, 65].
Mutations of the NLRP3 gene are found in almost 70% of patients with a CAPS phenotype.
IL-1β plays a pivotal role in the pathogenesis of many inflammatory conditions and represents a potential therapeutic target intervention in many inflammatory diseases. Growing knowledge of the pathogenic consequences of gene mutations involved in the monogenic autoinflammatory diseases has shed light on some pivotal pathophysiological mechanisms related to the activation and secretion of IL-1β.
Unlike most cytokines, IL-1β lacks a secretory signal peptide and is externalised by monocytic cells through a non-classical pathway, arranged in two steps. In the first one, toll-like receptor ligands, such as LPS, induce gene expression and synthesis of the inactive IL-1β precursor (pro-IL-1β). A second stimulus, such as exogenous ATP, strongly enhances IL-1β proteolytic maturation and secretion. ATP-triggered IL-1β secretion is mediated by P2X7 receptors expressed on the surface of monocytes; this mechanism is characterised by a series of events, only partially clarified, involving the secretory lysosomes.
Cryopyrin (NLRP3) is a key protein of the inflammasome. It is a member of the NOD-like receptor (NLR) protein family. In the presence of several stimuli, cryopyrin oligomerises and binds the adaptor protein ASC (Apoptosis associated Speck-like protein containing a CARD). This interaction directly activates two molecules of caspase-1 which, in turn, converts pro-IL-1β to the mature, active 17 kDa form (fig. 1). Thus activated cryopyrin induces the release of the active form IL-1β [66]. Experimental mouse models have revealed that monocytes from knockout mice deficient in cryopyrin cannot activate caspase-1 upon LPS and ATP stimulation, resulting in lack of IL-1β secretion. On the contrary, mutations in the cryopyrin gene in humans are associated with its gain of function leading to an enhanced and quicker production of IL-1β, even in the absence of a second signal. The speedier production and secretion of IL-1β in CAPS monocytes is probably linked to overactivation of their redox state [67].
FCAS is characterised by urticarial rash and fever spikes of short duration (usually <24 h) induced by cold exposure. Arthralgia and conjunctivitis are also common. Other symptoms observed following cold exposure include profuse sweating, drowsiness, headache, extreme thirst and nausea [68].
Muckle-Wells syndrome is characterised by recurrent episodes of urticaria (fig. 2b) and fever that may develop in early infancy. The fever episodes (usually below 38 °C) can be associated with the same clinical manifestations observed in FCAS (arthralgia, conjunctivitis, drowsiness), but usually are not strictly triggered by cold exposure. Acute phase reactants are elevated during fever episodes, and may also persist slightly increased during free intervals. During the course of the disease, neurosensorial deafness and polyarthritis may develop. Amyloid A (AA) amyloidosis is a complication of the late stage of the disease [62, 69, 70].
CINCA represents the more severe phenotype associated with mutations of the cryopyrin gene. An urticaria-like rash may develop during the first weeks of life. Many affected individuals present a typical “facies” characterised by frontal bossing, saddle back nose and midface hypoplasia, causing a sibling-like resemblance. Bone involvement is another hallmark of the disease. The most characteristic feature is represented by bony overgrowths predominantly involving the knees (including the patella) and the distal extremities of hands and feet. Major alterations in the organisation of cartilage growth have been described in biopsy specimens [71]. Chronic inflammatory polyarthritis may also be present, sometimes resulting in bone erosions [72]. Central nervous system (CNS) manifestations include chronic aseptic meningitis, increased intracranial pressure, cerebral atrophy, ventriculomegaly, sensorineural hearing loss and chronic papilloedema (fig. 2c), with associated optic-nerve atrophy and loss of vision. Mental retardation and seizures have also been reported. Patients exhibit persistent elevation of acute phase reactants, leucocytosis and chronic anaemia [63, 73–75].
Up to the present some 100 variants have been associated with either of the 3 phenotypic forms (http://fmf.igh.cnrs.fr/infevers/). Almost all the observed mutations are found in exon 3 of the NLRP3 gene, coding for the NACHT domain of cryopyrin that plays a crucial role in oligomerisation of the protein.
The pivotal role of cryopyrin in driving caspase 1 activation and the massive secretion of mature IL-1β observed in cryopyrin-mutated individuals suggested that anti-IL-1 treatment could represent an effective therapy. Initial isolated case reports suggested that the recombinant IL-1 receptor antagonist, anakinra, has a dramatic effect in controlling rash and constitutional symptoms of FCAS and Muckle-Wells patients [76]. These findings have been confirmed in different studies on CINCA patients [77].
Anakinra is given at a starting dosage of 1 mg/kg per day SC. Soon after the first injection all patients showed a dramatic improvement of urticarial rash, arthritis, headache and fever, with a complete resolution within one week from the beginning of the treatment A rapid decrease in acute phase reactants is also observed in the first weeks of treatment, with complete normalisation in the majority of patients. The same excellent results were recently observed using other IL-1 blockers such as IL-1 Trap (rilonacept) and anti-IL-1 monoclonal antibodies (canakinumab) [78–80]. Rilonacept has recently been approved for FCAS or MWS patients aged over 11 years and canakinumab for CAPS patients aged over 4 years.
The positive effects of anti-IL-1 blockers persist over the time. Monitoring of patients under anakinra treatment with a follow-up of over 3 years revealed that all CINCA patients still displayed complete control of the inflammatory symptoms with almost complete normalisation of the general conditions. Improvement of hearing loss after anakinra treatment has been described in almost 30% of CINCA patients [77].
| Table 1: The autoinflammatory diseases. | |||||
| Diseases | GeneChromosome | Protein | Transmission | Clinical features | |
| Periodic / recurrent fevers | Familial Mediterranean fever | MEVF 16p13.3 | Pyrin | AR | Short duration of fever episodes: 24–48 hours. Abdominal and chest pain. Erysipelas-like erythema. High incidence of renal amyloidosis in untreated patients. Good response to colchicine. Possible response to IL-1 blockade. |
| Mevalonate kinase deficiency | MVK12q24 | Mevalonate kinase | AR | Early onset (usually <12 months). Mean duration of fever episodes: 4–5 days. Poor conditions during fever episodes. Abdominal pain, vomiting and diarrhoea. Splenomegaly. Good response to steroids. High rate of self-resolution during adulthood. Amyloidosis is rare. | |
| TNF receptor associated periodic syndrome | TNFRSF1A12p13 | p55 TNF receptor | AD | Prolonged fever episodes: 1–3 weeks. Periorbital oedema, monocytic fasciitis. Incidence of renal amyloidosis: 15‒25%. Response to TNF- and IL-1 blockade. | |
| NALP12-associated periodic Fever | NALP12 19q13 | NALP12 | AD | Periodic fever after cold exposure, hearing loss. | |
| Cryopyrinopathies | FCAS, MWS, CINCA | CIAS 1/NALP31q44 | Cryopyrin | AD | FCAS: rash, fever and arthralgia after cold exposure. MWS: recurrent or sub-chronic urticaria-like lesions, sensorineural hearing loss, amyloidosis. CINCA: as above + mental retardation, chronic aseptic meningitis and bone deformities. Good response to IL-1 blockade. |
| Granulomatous disorders | Blau’s syndrome | CARD15/NOD216q12 | CARD15 | AD | Early onset (<5 years). Polyarticular granulomatous arthritis, uveitis, skin rash. Good response to anti-TNF monoclonal antibodies. |
| Pyogenic disorders | PAPA syndrome | PSTPIP115q24-q25.1 | PSTPIP1 | AD | Pyogenic sterile arthritis, pyogenic gangrenosum, cystic acne. Good response to IL-1 blockade. |
| Majeed’s syndrome DIRA | LPIN218p IL1RN2q | LPIN2 IL-1 receptor antagonist | AR AR | Multifocal osteomyelitis, congenital dyserythropoietic anaemia, inflammatory dermatosis. Neonatal-onset multifocal osteomyelitis, periostitis, and pustulosis. Dramatic response to anakinra. | |
| FCAS = Familial cold autoinflammatory syndrome; MWS = Muckle-Wells syndrome; CINCA = Chronic infantile neurological cutaneous and articular syndrome; PAPA = Pyogenic sterile arthritis, pyoderma gangrenosum and acne (PAPA) syndrome; CRMO = Chronic recurrent multifocal osteomyelitis; DIRA = Deficiency of the interleukin-1-receptor antagonist; AR = autosomal recessive; AD = autosomal dominant. *Identification of the gene defect. | |||||
This disease was originally identified in two families originating from Guadeloupe, who presented a clinical picture involving recurrent fever and cold sensitivity associated with some of the following additional symptoms: neuronal hearing loss, aphthous ulcers, lymphadenopathy, abdominal pain and acute phase response. Using a candidate gene approach, mutations in another member of an NLR family (the same of NLRP3) was found. The affected members of the two families were indeed carriers of non-ambiguous mutations (i.e. nonsense and splice site) of the NLRP12gene. Evaluation of other additional families confirmed the relatively benign nature of this disorder, which is essentially featured by transitory occurrence of urticarial rash, muscle-skeletal pain and general discomfort following exposure to some environmental trigger factors, such as cold or fatigue.
Even if NLRP12 has been shown to play a role in the regulation of the pro-inflammatory NF-κB pathway [81], accelerated secretion of IL-1 secondary to a deregulated redox state has been suggested as an alternative pathogenic mechanism [82].
Blau syndrome, or familial juvenile systemic granulomatosis, is an autosomal-dominant, autoinflammatory disease characterised by a noncaseating granulomatous inflammation affecting the joint, the skin, and the uveal tract (the triad of arthritis, dermatitis, and uveitis) [83]. The gene responsible for Blau syndrome, NOD2/CARD15, encodes a protein containing a NACHT domain [84]. To date more than 10 different genetic mutations leading to substitutions in or near the NACHT domain of NOD2/CARD15 have been documented in affected patients with either the familial or the sporadic presentation [84–86].
NOD2/CARD15 belongs to the superfamily of NOD (nucleotide oligomerisation domain)-like receptors (NLR), that are intracellular receptors for bacterial peptidoglycans. NOD2/CARD15 recognises muramyl dipeptide (MDP), the minimal motif of peptidoglycan of both Gram-positive and Gram-negative bacteria. After stimulation with MDP, NOD2/CARD15 is able to induce both NF-κB activation and release of bioactive IL-1β in a caspase 1-dependent manner. It is, therefore, conceivable that in Blau syndrome the mutation of the NACHT domain causes a gain of the protein’s function resulting in a sustained pro-inflammatory state.
Disease onset is usually observed during the first years of life. A symmetrical polyarticular arthritis with a “boggy” appearance is the typical joint manifestation (fig. 2d) and is mainly due to a exuberant tenosynovitis. Eye involvement is characterised by intermediate uveitis or panuveitis. 50% of the patients with ocular involvement develop cataracts, and approximately one-third may evolve into secondary glaucoma. A typical tan-coloured, scaly, ichthyosiform rash is seen in almost 90% of the affected individuals [87, 88]. Patients are treated with oral steroids and immunosuppressive drugs (methotrexate, cyclosporin A) with variable outcomes. Recent anecdotal reports suggest a beneficial effect of anti-TNF (infliximab) [87] and anti-IL-1 treatment [88].
Pyogenic sterile arthritis, pyoderma gangrenosum and acne syndrome (PAPA, MIM 604416) is a disorder caused by mutations of gene coding for the CD2-binding protein 1 (CD2BP1), or PSTPIP1[89, 90]. The manifestations of this disorder are pyogenic gangrenosum, cystic acne, and pyogenic sterile arthritis which represents the most common symptom of the disease. The arthritis usually has its onset in early childhood. It is pauciarticular in nature, affecting one to three joints at a time, and is characterised by recurring inflammatory episodes that resemble septic arthritis (fig. 2e) and lead to accumulation of pyogenic, neutrophil-rich material within the affected joints, which ultimately results in significant synovial and cartilage destruction [89–91]. Cultures of the skin lesions and joint fluid of these patients are sterile. Dermatological manifestations are also episodic and recurrent, with onset usually during the second decade of life, and are characterised by debilitating, aggressive, ulcerative skin lesions, usually of the lower extremities, indistinguishable from pyogenic gangrenosum. Sterile abscesses at injection sites may also be observed.
Positional cloning in informative families identified two mis-sense mutations within the CD2BP1 (or PSTPIP1) gene(90). PSTPIP1 has been shown to bind pyrin [92]. It has been postulated that mutated PSTPIP1 displays an higher affinity for binding with pyrin, leading to increased susceptibility to inflammation.
PAPA syndrome has been reported to be generally responsive to oral glucocorticoids [89, 90]. However, sustained remission in a steroid-resistant patient and a consistent amelioration of the cutaneous manifestations have been anecdotally reported after anti-TNF [93, 94] and anti-IL-1 treatment [95].
In 1989 three related Arab children presenting an association of chronic recurrent multifocal osteomyelitis (CRMO), and inflammatory dermatosis were described by Majeed and co-workers [96].
Unlike isolated CRMO, bone manifestations have an earlier age at onset, present with a higher frequency of recurrence and show shorter and less frequent remissions. Congenital dyserythropoietic anaemia is characterised by microcytosis both peripherally and in the bone marrow, and the need of repeated blood transfusions may be required. Inflammatory dermatosis may vary from Sweet syndrome to chronic pustulosis. Recurrent fever episodes and growth failure have also been reported [96, 97]. Non-steroidal anti-inflammatory drugs are moderately helpful, whereas short courses of oral steroids promptly control disease relapses, even if steroid-dependence has been reported. Colchicine does not seem to be effective [97]. Splenectomy has reportedly been able to control the haematological manifestations [97].
In 2005 a linkage analysis was performed on two new Jordanian families allowing identification of the LPIN2 gene mapped on chromosome 18p [98].
The existence of a monogenic disease associated with CRMO has raised the hypothesis that sporadic CRMO could also be included in the group of autoinflammatory diseases. This theory has been further supported by the recent identification on a chromosome 18 of a mis-sense mutation in the PSTPIP2 gene in a spontaneous murine model of an autoinflammatory disorder characterised by chronic multifocal osteomyelitis [99].
DIRA is a recently identified autosomal recessive autoinflammatory syndrome, due to the deficiency of the interleukin-1-receptor antagonist, which begins around birth with multifocal osteomyelitis, periostitis, and pustulosis. Persistent elevation of acute phase reactants (ESR and CRP) are observed from birth. The skin manifestations range from groupings of small pustules to a generalised pustulosis. The bone manifestations include osteolytic lesions with a sclerotic rim, epiphyseal ballooning of multiple distal and proximal long bones, widening of ribs and clavicles, heterotopic ossification or periosteal cloaking of the proximal femoral metaphysis and periosteal elevation of the diaphysis [100]. The patients so far described exhibit homozygous truncating mutations in the IL1RN gene. As a result of these mutations, there is no secretion of interleukin-1-receptor antagonist (IL1RA) protein, which usually inhibits the proinflammatory cytokines interleukin-1 including interleukin-1β. Deficiency of IL1RA being causative of the disease, these patients show a dramatic response to the substitutive treatment with recombinant IL-1 receptor antagonist (anakinra) [100].
Several multi-factorial inflammatory diseases present clinical similarities to inherited autoinflammatory diseases and are thought to be mainly autoinflammatory in nature [101]. Some of these disorders have also shown a bright response to IL-1 inhibitors.
The PFAPA (Periodic Fever, Aphthous stomatitis, Pharyngitis and Adenitis) syndrome is characterised by periodic fever attacks similar to those observed in monogenic periodic fevers in children negative for mutations of MEFV, MVK and TNFRS1A genes. In fact, although some anecdotal familial cases of PFAPA have been reported, a genetic basis has never been demonstrated.
It was first described by Marshall et al. in 1987 [98].
It is characterised by regularly (often clockwork) recurrent episodes of fever lasting 3–6 days. The frequency of episodes varies but they usually present every 2–6 weeks. The diagnosis is based on clinical criteria that require the presence of a disease onset before the age of 5 years and at least one of the three associated constitutive symptoms (aphthosis, cervical adenitis, pharyngitis) in the absence of upper respiratory tract infection or cyclic neutropenia [102].
Pharyngitis is the most frequent and characteristic symptom associated with fever attacks. It is erythematous or exsudative, and self-limiting. The throat swab is always negative. The stomatitis is characterised by small lesions that may appear some days before the occurrence of fever and present with a spontaneous self- remission. The cervical lymph nodes are usually tender and enlarged during fever episodes, with subsequent gradual normalisation with resolution of the fever attack. Acute phase reactants and neutrophils are elevated during the attacks and normalise completely during the periods of complete wellbeing.
The disease runs a benign course and tends to remit spontaneously with time. Typically a single administration of oral steroids at the beginning of the episode promptly aborts the fever attack; however, this treatment is reported to shorten the disease-free intervals [103].
Children with PFAPA syndrome often appear in good condition during the fever spikes as well. This is a clinical feature that may help to distinguish PFAPA from the other form of periodic fever associated with a genetic defect.
The diagnostic criteria for PFAPA syndrome have been shown to have low specificity.
In fact, almost 50% of genetically positive children (especially MKD) also fulfill the PFAPA criteria, which, therefore, do not represent a specific tool capable of selecting patients with strong probability of being negative at genetic testing. In this line, some clinical manifestations during fever attacks, such as abdominal and chest pain and gastrointestinal symptoms (vomiting and/or diarrhoea), should be considered evocative for a higher risk of carrying mutations of known genes [104, 105].
Gout and pseudogout (two common adult rheumatic diseases) are caused respectively by deposition of monosodium urate (MSU) and calcium pyrophosphate dihydrate (CPPD) crystals in the joints and periarticular tissues. It has been shown that both MSU and CPPD crystals are able to activate the inflammasome [106]. For this reason IL-1 blockade has been used in colchicine-resistant gout with a good response [107].
Another disease sharing a number of clinical features (systemic inflammation, arthritis, rash, persistent elevation of acute phase reactants) with inherited autoinflammatory diseases is systemic onset juvenile idiopathic arthritis (SoJIA).
In some studies, gene expression analysis revealed the presence of a prevalent IL-1β signature in SoJIA [108, 109] and a variable percentage of these patients (from 40 to 87% according to the different studies) show a dramatic and persistent response to anti-IL-1 blockade similar to that observed in CAPS patients [108, 110].
Idiopathic recurrent pericarditis too presents many features that are consistent with an autoinflammatory disease. A recent study described 3 steroid-dependent and colchicine-resistant children with recurrent pericarditis treated by anakinra with a dramatic response. The author reports that pericarditis recurred when anakinra treatment was discontinued, and that no further episodes occurred after it was resumed [111].
Finally, recent evidence has shown that the same pathogenic mechanisms responsible for the activation of innate immunity in inherited autoinflammatory diseases may also play a crucial role in sustaining inflammation in several extremely frequent multi-factorial illnesses, such as type II diabetes [112] and atherosclerosis [113], opening new perspectives on the management of these diseases.
1 Siegal S. Benign paroxysmal peritonitis. Ann Intern Med. 1945;23:1–21.
2 Ben-Chetrit E, Touitou I. Familial Mediterranean fever in the world. Arthritis Rheum. 2009;61(10):1447–53.
3 The French FMF Consortium. A candidate gene for familial Mediterranean fever. Nat Genet. 1997;17(1):25–31.
4 The International FMF Consortium. Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell. 1997;90(4):797–807.
5 Chae JJ, Komarow HD, Cheng J, Wood G, Raben N, Liu PP, et al. Targeted disruption of pyrin, the FMF protein, causes heightened sensitivity to endotoxin and a defect in macrophage apoptosis. Mol Cell. 2003;11(3):591–604.
6 Dinarello CA. A signal for the caspase-1 inflammasome free of TLR. Immunity. 2007;26(4):383–5.
7 Chae JJ, Wood G, Richard K, Jaffe H, Colburn NT, Masters SL, et al. The familial Mediterranean fever protein, pyrin, is cleaved by caspase-1 and activates NF-kappa B through its N-terminal fragment. Blood. 2008;112(5):1794–803.
8 Chae JJ, Cho YH, Lee GS, Cheng J, Liu PP, Feigenbaum L, et al. Gain-of-function pyrin mutations induce NLRP3 protein-independent interleukin-1beta activation and severe autoinflammation in mice. Immunity. 2011;34(5):755–68.
9 La Regina M, Nucera G, Diaco M, Procopio A, Gasbarrini G, Notarnicola C, et al. Familial Mediterranean fever is no longer a rare disease in Italy (vol 11, pg 550, 2003). Eur J Hum Genet. 2003;11(7):550.
10 Tunca M, Akar S, Onen F, Ozdogan H, Kasapcopur O, Yalcinkaya F, et al. Familial Mediterranean fever (FMF) in Turkey: results of a nationwide multicenter study. Medicine. (Baltimore) 2005;84(1):1–11.
11 Gedalia A, Adar A, Gorodischer R. Familial Mediterranean fever in children. J Rheumatol Suppl. 1992;35:1–9.
12 Livneh A, Langevitz P, Zemer D, Zaks N, Kees S, Lidar T, et al. Criteria for the diagnosis of familial Mediterranean fever. Arthritis Rheum. 1997;40(10):1879–85.
13 Yalcinkaya F, Ozen S, Ozcakar ZB, Aktay N, Cakar N, Duzova A, et al. A new set of criteria for the diagnosis of familial Mediterranean fever in childhood. Rheumatology. (Oxford) 2009;48(4):395–8.
14 Ben-Zvi I, Brandt B, Berkun Y, Lidar M, Livneh A. The relative contribution of environmental and genetic factors to phenotypic variation in familial Mediterranean fever (FMF). Gene. 2012;491(2):260–3.
15 Lachmann HJ, Goodman HJ, Gilbertson JA, Gallimore JR, Sabin CA, Gillmore JD, et al. Natural history and outcome in systemic AA amyloidosis. N Engl J Med. 2007;356(23):2361–71.
16 Touitou I, Sarkisian T, Medlej-Hashim M, Tunca M, Livneh A, Cattan D, et al. Country as the primary risk factor for renal amyloidosis in familial Mediterranean fever. Arthritis Rheum. 2007;56(5):1706–12.
17 Bakkaloglu A, Duzova A, Ozen S, Balci B, Besbas N, Topaloglu R, et al. Influence of Serum Amyloid A (SAA1) and SAA2 gene polymorphisms on renal amyloidosis, and on SAA/C-reactive protein values in patients with familial mediterranean fever in the Turkish population. J Rheumatol. 2004;31(6):1139–42.
18 Goldfinger SE. Colchicine for familial Mediterranean fever. N Engl J Med. 1972;287(25):1302.
19 Bakkaloglu A. Familial Mediterranean fever. Pediatr Nephrol. 2003;18(9):853–9.
20 Ozkaya N, Yalcinkaya F. Colchicine treatment in children with familial Mediterranean fever. Clin Rheumatol. 2003;22(4–5):314–7.
21 Aybal KA, Yildirim T, Altindal M, Arici M, Yasavul U, Turgan C. AA type renal amyloidosis secondary to FMF: a long-term follow-up in two patients. Ren Fail. 2010;32(10):1230–2.
22 van der Meer JW, Vossen JM, Radl J, van Nieuwkoop JA, Meyer CJ, Lobatto S, et al. Hyperimmunoglobulinaemia D and periodic fever: a new syndrome. Lancet. 1984;1(8386):1087–90.
23 Gibson KM, Hoffmann GF, Tanaka RD, Bishop RW, Chambliss KL. Mevalonate kinase map position 12q24. Chromosome Res. 1997;5(2):150.
24 D’Osualdo A, Picco P, Caroli F, Gattorno M, Giacchino R, Fortini P, et al. MVK mutations and associated clinical features in Italian patients affected with autoinflammatory disorders and recurrent fever. Eur J Hum Genet. 2005;13(3):314–20.
25 Lawrence A, Hol F, Aggarwal A, Drenth JP. Hyperimmunoglobulinaemia D syndrome in India: report of two siblings with a novel mutation. Ann Rheum Dis. 2006;65(12):1674–6.
26 Houten SM, Kuis W, Duran M, De Koning TJ, van Royen-Kerkhof A, Romeijn GJ, et al. Mutations in MVK, encoding mevalonate kinase, cause hyperimmunoglobulinaemia D and periodic fever syndrome. Nat Genet. 1999;22(2):175–7.
27 Cuisset L, Drenth JP, Simon A, Vincent MF, van dV, V, van der Meer JW, et al. Molecular analysis of MVK mutations and enzymatic activity in hyper-IgD and periodic fever syndrome. Eur J Hum Genet. 2001;9(4):260–6.
28 Hoffmann GF, Charpentier C, Mayatepek E, Mancini J, Leichsenring M, Gibson KM, et al. Clinical and biochemical phenotype in 11 patients with mevalonic aciduria. Pediatrics. 1993;91(5):915–21.
29 Mandey SH, Kuijk LM, Frenkel J, Waterham HR. A role for geranylgeranylation in interleukin-1beta secretion. Arthritis Rheum. 2006;54(11):3690–5.
30 Drenth JP, Haagsma CJ, van der Meer JW. Hyperimmunoglobulinemia D and periodic fever syndrome. The clinical spectrum in a series of 50 patients. International Hyper-IgD Study Group. Medicine. (Baltimore) 1994;73(3):133–44.
31 Frenkel J, Houten SM, Waterham HR, Wanders RJ, Rijkers GT, Duran M, et al. Clinical and molecular variability in childhood periodic fever with hyperimmunoglobulinaemia D. Rheumatology. (Oxford) 2001;40(5):579–84.
32 Obici L, Manno C, Muda AO, Picco P, D’Osualdo A, Palladini G, et al. First report of systemic reactive (AA) amyloidosis in a patient with the hyperimmunoglobulinemia D with periodic fever syndrome. Arthritis Rheum. 2004;50(9):2966–9.
33 Ammouri W, Cuisset L, Rouaghe S, Rolland MO, Delpech M, Grateau G, et al. Diagnostic value of serum immunoglobulinaemia D level in patients with a clinical suspicion of hyper IgD syndrome. Rheumatology. (Oxford) 2007;46(10):1597–600.
34 Saulsbury FT. Hyperimmunoglobulinemia D and periodic fever syndrome (HIDS) in a child with normal serum IgD, but increased serum IgA concentration. J Pediatr. 2003;143(1):127–9.
35 Touitou I, Lesage S, McDermott M, Cuisset L, Hoffman H, Dode C, et al. Infevers: An evolving mutation database for auto-inflammatory syndromes. Hum Mutat. 2004;24(3):194–8.
36 Mandey SH, Schneiders MS, Koster J, Waterham HR. Mutational spectrum and genotype-phenotype correlations in mevalonate kinase deficiency. Hum Mutat. 2006;27(8):796–802.
37 Simon A, Kremer HP, Wevers RA, Scheffer H, De Jong JG, van der Meer JW, et al. Mevalonate kinase deficiency: Evidence for a phenotypic continuum. Neurology. 2004;62(6):994–7.
38 Drenth JP, Vonk AG, Simon A, Powell R, van der Meer JW. Limited efficacy of thalidomide in the treatment of febrile attacks of the hyper-IgD and periodic fever syndrome: a randomized, double-blind, placebo-controlled trial. J Pharmacol Exp Ther. 2001;298(3):1221–6.
39 Takada K, Aksentijevich I, Mahadevan V, Dean JA, Kelley RI, Kastner DL. Favorable preliminary experience with etanercept in two patients with the hyperimmunoglobulinemia D and periodic fever syndrome. Arthritis Rheum. 2003;48(9):2645–51.
40 Marchetti F, Barbi E, Tommasini A, Oretti C, Ventura A. Inefficacy of etanercept in a child with hyper-IgD syndrome and periodic fever. Clin Exp Rheumatol. 2004;22(6):791–2.
41 Cailliez M, Garaix F, Rousset-Rouviere C, Bruno D, Kone-Paut I, Sarles J, et al. Anakinra is safe and effective in controlling hyperimmunoglobulinaemia D syndrome-associated febrile crisis. J Inherit Metab Dis. 2006;29(6):763.
42 McDermott MF, Aksentijevich I, Galon J, McDermott EM, Ogunkolade BW, Centola M, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell. 1999;97(1):133–44.
43 Williamson LM, Hull D, Mehta R, Reeves WG, Robinson BH, Toghill PJ. Familial Hibernian fever. Q J Med. 1982;51(204):469–80.
44 Aksentijevich I, Galon J, Soares M, Mansfield E, Hull K, Oh HH, et al. The tumor-necrosis-factor receptor-associated periodic syndrome: new mutations in TNFRSF1A, ancestral origins, genotype-phenotype studies, and evidence for further genetic heterogeneity of periodic fevers. Am J Hum Genet. 2001;69(2):301–14.
45 Aganna E, Hammond L, Hawkins PN, Aldea A, McKee SA, van Amstel HK, et al. Heterogeneity among patients with tumor necrosis factor receptor-associated periodic syndrome phenotypes. Arthritis Rheum. 2003;48(9):2632–44.
46 D’Osualdo A, Ferlito F, Prigione I, Obici L, Meini A, Zulian F, et al. Neutrophils from patients with TNFRSF1A mutations display resistance to tumor necrosis factor-induced apoptosis – Pathogenetic and clinical implications. Arthritis Rheum. 2006;54(3):998–1008.
47 Ravet N, Rouaghe S, Dode C, Bienvenu J, Stirnemann J, Levy P, et al. Clinical significance of P46L and R92Q substitutions in the tumour necrosis factor superfamily 1A gene. Ann Rheum Dis. 2006;65(9):1158–62.
48 Siebert S, Amos N, Fielding CA, Wang EC, Aksentijevich I, Williams BD, et al. Reduced tumor necrosis factor signaling in primary human fibroblasts containing a tumor necrosis factor receptor superfamily 1A mutant. Arthritis Rheum. 2005;52(4):1287–92.
49 Bulua AC, Simon A, Maddipati R, Pelletier M, Park H, Kim KY, et al. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J Exp Med. 2011;208(3):519–33.
50 Pelagatti MA, Meini A, Caorsi R, Cattalini M, Federici S, Zulian F, et al. Long-term clinical profile of children with the low-penetrance R92Q mutation of the TNFRSF1A gene. Arthritis Rheum. 2011;63(4):1141–50.
51 Hull KM, Drewe E, Aksentijevich I, Singh HK, Wong K, McDermott EM, et al. The TNF receptor-associated periodic syndrome (TRAPS): emerging concepts of an autoinflammatory disorder. Medicine. (Baltimore) 2002;81(5):349–68.
52 Drewe E, McDermott EM, Powell PT, Isaacs JD, Powell RJ. Prospective study of anti-tumour necrosis factor receptor superfamily 1B fusion protein, and case study of anti-tumour necrosis factor receptor superfamily 1A fusion protein, in tumour necrosis factor receptor associated periodic syndrome (TRAPS): clinical and laboratory findings in a series of seven patients. Rheumatology. 2003;42(2):235–9.
53 Kallinich T, Briese S, Roesler J, Rudolph B, Sarioglu N, Blankenstein O, et al. Two familial cases with tumor necrosis factor receptor-associated periodic syndrome caused by a non-cysteine mutation (T50M) in the TNFRSF1A gene associated with severe multiorganic amyloidosis. J Rheumatol. 2004;31(12):2519–22.
54 Weyhreter H, Schwartz M, Kristensen TD, Valerius NH, Paerregaard A. A new mutation causing autosomal dominant periodic fever syndrome in a Danish family. J Pediatr. 2003;142(2):191–3.
55 Simon A, Bodar EJ, van der Hilst JC, van der Meer JW, Fiselier TJ, Cuppen MP, et al. Beneficial response to interleukin 1 receptor antagonist in traps. Am J Med. 2004;117(3):208–10.
56 Arostegui JI, Solis P, Aldea A, Cantero T, Rius J, Bahillo P, et al. Etanercept plus colchicine treatment in a child with tumour necrosis factor receptor-associated periodic syndrome abolishes auto-inflammatory episodes without normalising the subclinical acute phase response. Eur J Pediatr. 2005;164(1):13–6.
57 Jacobelli S, Andre M, Alexandra JF, Dode C, Papo T. Failure of anti-TNF therapy in TNF Receptor 1-Associated Periodic Syndrome (TRAPS). Rheumatology. (Oxford) 2007.
58 Bulua AC, Mogul DB, Aksentijevich I, Singh H, He D, Muenz L, et al. Efficacy of etanercept in the tumor necrosis factor receptor-associated periodic syndrome (TRAPS). Arthritis Rheum. 2011.
59 Nedjai B, Hitman GA, Quillinan N, Coughlan RJ, Church L, McDermott MF, et al. Proinflammatory action of the antiinflammatory drug infliximab in tumor necrosis factor receptor-associated periodic syndrome. Arthritis Rheum. 2009;60(2):619–25.
60 Gattorno M, Pelagatti MA, Meini A, Obici L, Barcellona R, Federici S, et al. Persistent efficacy of anakinra in patients with tumor necrosis factor receptor-associated periodic syndrome. Arthritis Rheum. 2008;58(5):1516–20.
61 Kile RL, Rusk HA. A case of cold urticaria with an unusual family history. JAMA. 1940;(114):1067–8.
62 Muckle TJ, WELLSM. Urticaria, deafness, and amyloidosis: a new heredo-familial syndrome. Q J Med. 1962;31:235–48.
63 Prieur AM, Griscelli C. Arthropathy with rash, chronic meningitis, eye lesions, and mental retardation. J Pediatr. 1981;99(1):79–83.
64 Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet. 2001;29(3):301–5.
65 Feldmann J, Prieur AM, Quartier P, Berquin P, Certain S, Cortis E, et al. Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1, a gene highly expressed in polymorphonuclear cells and chondrocytes. Am J Hum Genet. 2002;71(1):198–203.
66 Dinarello CA. Mutations in cryopyrin: bypassing roadblocks in the caspase 1 inflammasome for interleukin-1beta secretion and disease activity. Arthritis Rheum. 2007;56(9):2817–22.
67 Tassi S, Carta S, Vene R, Delfino L, Ciriolo MR, Rubartelli A. Pathogen-induced interleukin-1beta processing and secretion is regulated by a biphasic redox response. J Immunol. 2009;183(2):1456–62.
68 Hoffman HM, Wanderer AA, Broide DH. Familial cold autoinflammatory syndrome: phenotype and genotype of an autosomal dominant periodic fever. J Allergy Clin Immunol. 2001;108(4):615–20.
69 Nazzari G, Desirello G, Crovato F. Recurrent urticarial skin eruption since infancy. Muckle-Wells syndrome (MWS). Arch Dermatol. 1995;131(1):81–5.
70 Leslie KS, Lachmann HJ, Bruning E, McGrath JA, Bybee A, Gallimore JR, et al. Phenotype, genotype, and sustained response to anakinra in 22 patients with autoinflammatory disease associated with CIAS-1/NALP3 mutations. Arch Dermatol. 2006;142(12):1591–7.
71 De Cunto CL, Liberatore DI, San Roman JL, Goldberg JC, Morandi AA, Feldman G. Infantile-onset multisystem inflammatory disease: a differential diagnosis of systemic juvenile rheumatoid arthritis. J Pediatr. 1997;130(4):551–6.
72 Lequerre T, Vittecoq O, Saugier-Veber P, Goldenberg A, Patoz P, Frebourg T, et al. A cryopyrin-associated periodic syndrome with joint destruction. Rheumatology. (Oxford) 2007;46(4):709–14.
73 Prieur AM, Griscelli C, Lampert F, Truckenbrodt H, Guggenheim MA, Lovell DJ, et al. A chronic, infantile, neurological, cutaneous and articular (CINCA) syndrome. A specific entity analysed in 30 patients. Scand J Rheumatol Suppl. 1987;66:57–68.
74 Aksentijevich I, Nowak M, Mallah M, Chae JJ, Watford WT, Hofmann SR, et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum. 2002;46(12):3340–8.
75 Caroli F, Pontillo A, D’Osualdo A, Travan L, Ceccherini I, Crovella S, et al. Clinical and genetic characterization of Italian patients affected by CINCA syndrome. Rheumatology. (Oxford) 2007;46(3):473–8.
76 Hawkins PN, Lachmann HJ, McDermott MF. Interleukin-1-receptor antagonist in the Muckle-Wells syndrome. N Engl J Med. 2003;348(25):2583–4.
77 Goldbach-Mansky R, Dailey NJ, Canna SW, Gelabert A, Jones J, Rubin BI, et al. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. N Engl J Med. 2006;355(6):581–92.
78 Kuemmerle-Deschner JB, Tzaribachev N, Hausmann S, Gramlich K, Felix SD, az-Lorente M, et al. Long-lasting response to ACZ885 (a new human IgG1 anti-IL-1 beta monoclonal antibody) in patients with Muckle-Wells Syndrome (MWS). Clinical and Experimental Rheumatology. 2008;26(2):180.
79 Lachmann H, Jung T, Felix S, Lowe P, Offer M, Rordorf C, et al. Treatment of cryopyrin associated periodic fever syndrome with a fully human anti-IL-1beta monoclonal antibody (ACZ885): results from a subcutaneous administration study. Clin Exp Rheumatol. 2008;26(2):181.
80 Lachmann HJ, Kone-Paut I, Kuemmerle-Deschner JB, Leslie KS, Hachulla E, Quartier P, et al. Use of canakinumab in the cryopyrin-associated periodic syndrome. N Engl J Med. 2009;360(23):2416–25.
81 Jeru I, Duquesnoy P, Fernandes-Alnemri T, Cochet E, Yu JW, Lackmy-Port-Lis M, et al. Mutations in NALP12 cause hereditary periodic fever syndromes. Proc Natl Acad Sci U S A. 2008;105(5):1614–9.
82 Borghini S, Tassi S, Chiesa S, Caroli F, Carta S, Caorsi R, et al. Clinical presentation and pathogenesis of cold-induced autoinflammatory disease in a family with recurrence of a NLRP12 mutation. Arthritis Rheum. 2010.
83 Blau EB. Familial granulomatous arthritis, iritis, and rash. J Pediatr. 1985;107(5):689–93.
84 Miceli-Richard C, Lesage S, Rybojad M, Prieur AM, Manouvrier-Hanu S, Hafner R, et al. CARD15 mutations in Blau syndrome. Nat Genet. 2001;29(1):19–20.
85 Kanazawa N, Okafuji I, Kambe N, Nishikomori R, Nakata-Hizume M, Nagai S, et al. Early-onset sarcoidosis and CARD15 mutations with constitutive nuclear factor-kappaB activation: common genetic etiology with Blau syndrome. Blood. 2005;105(3):1195–7.
86 van Duist MM, Albrecht M, Podswiadek M, Giachino D, Lengauer T, Punzi L, et al. A new CARD15 mutation in Blau syndrome. Eur J Hum Genet. 2005;13(6):742–7.
87 Rose CD, Wouters CH, Meiorin S, Doyle TM, Davey MP, Rosenbaum JT, et al. Pediatric granulomatous arthritis: an international registry. Arthritis Rheum. 2006;54(10):3337–44.
88 Arostegui JI, Arnal C, Merino R, Modesto C, Antonia CM, Moreno P, et al. NOD2 gene-associated pediatric granulomatous arthritis: clinical diversity, novel and recurrent mutations, and evidence of clinical improvement with interleukin-1 blockade in a Spanish cohort. Arthritis Rheum. 2007;56(11):3805–13.
89 Lindor NM, Arsenault TM, Solomon H, Seidman CE, McEvoy MT. A new autosomal dominant disorder of pyogenic sterile arthritis, pyoderma gangrenosum, and acne: PAPA syndrome. Mayo Clin Proc. 1997;72(7):611–5.
90 Wise CA, Gillum JD, Seidman CE, Lindor NM, Veile R, Bashiardes S, et al. Mutations in CD2BP1 disrupt binding to PTP PEST and are responsible for PAPA syndrome, an autoinflammatory disorder. Hum Mol Genet. 2002;11(8):961–9.
91 Yeon HB, Lindor NM, Seidman JG, Seidman CE. Pyogenic arthritis, pyoderma gangrenosum, and acne syndrome maps to chromosome 15q. Am J Hum Genet. 2000;66(4):1443–8.
92 Shoham NG, Centola M, Mansfield E, Hull KM, Wood G, Wise CA, et al. Pyrin binds the PSTPIP1/CD2BP1 protein, defining familial Mediterranean fever and PAPA syndrome as disorders in the same pathway. Proc Natl Acad Sci U S A. 2003;100(23):13501–6.
93 Cortis E, de BF, Insalaco A, Cioschi S, Muratori F, D’Urbano LE, et al. Abnormal production of tumor necrosis factor (TNF) – alpha and clinical efficacy of the TNF inhibitor etanercept in a patient with PAPA syndrome [corrected]. J Pediatr. 2004;145(6):851–5.
94 Stichweh DS, Punaro M, Pascual V. Dramatic improvement of pyoderma gangrenosum with infliximab in a patient with PAPA syndrome. Pediatr Dermatol. 2005;22(3):262–5.
95 Dierselhuis MP, Frenkel J, Wulffraat NM, Boelens JJ. Anakinra for flares of pyogenic arthritis in PAPA syndrome. Rheumatology. (Oxford) 2005;44(3):406–8.
96 Majeed HA, Kalaawi M, Mohanty D, Teebi AS, Tunjekar MF, al-Gharbawy F, et al. Congenital dyserythropoietic anemia and chronic recurrent multifocal osteomyelitis in three related children and the association with Sweet syndrome in two siblings. J Pediatr. 1989;115(5Pt 1):730–4.
97 Majeed HA, Al-Tarawna M, El-Shanti H, Kamel B, Al-Khalaileh F. The syndrome of chronic recurrent multifocal osteomyelitis and congenital dyserythropoietic anaemia. Report of a new family and a review. Eur J Pediatr. 2001;160(12):705–10.
98 Ferguson PJ, Chen S, Tayeh MK, Ochoa L, Leal SM, Pelet A, et al. Homozygous mutations in LPIN2 are responsible for the syndrome of chronic recurrent multifocal osteomyelitis and congenital dyserythropoietic anaemia (Majeed syndrome). J Med Genet. 2005;42(7):551–7.
99 Ferguson PJ, Bing X, Vasef MA, Ochoa LA, Mahgoub A, Waldschmidt TJ, et al. A missense mutation in pstpip2 is associated with the murine autoinflammatory disorder chronic multifocal osteomyelitis. Bone. 2006;38(1):41–7.
100 Aksentijevich I, Masters SL, Ferguson PJ, Dancey P, Frenkel J, van Royen-Kerkhoff A, et al. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N Engl J Med. 2009;360(23):2426–37.
101 Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease (*). Annu Rev Immunol. 2009;27:621–68.
102 Thomas KT, Feder HM, Lawton AR, Edwards KM. Periodic fever syndrome in children. J Pediatr. 1999;135(1):15–21.
103 Padeh S, Brezniak N, Zemer D, Pras E, Livneh A, Langevitz P, et al. Periodic fever, aphthous stomatitis, pharyngitis, and adenopathy syndrome: clinical characteristics and outcome. J Pediatr. 1999;135(1):98–101.
104 Gattorno M, Sormani MP, D’Osualdo A, Pelagatti MA, Caroli F, Federici S, et al. A diagnostic score for molecular analysis of hereditary autoinflammatory syndromes with periodic fever in children. Arthritis Rheum. 2008;58(6):1823–32.
105 Gattorno M, Caorsi R, Meini A, Cattalini M, Federici S, Zulian F, et al. Differentiating PFAPA syndrome from monogenic periodic fevers. Pediatrics. 2009;124(4):e721–e728.
106 Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440(7081):237–41.
107 McGonagle D, Tan AL, Shankaranarayana S, Madden J, Emery P, McDermott MF. Management of treatment resistant inflammation of acute on chronic tophaceous gout with anakinra. Ann Rheum Dis. 2007;66(12):1683–4.
108 Pascual V, Allantaz F, Arce E, Punaro M, Banchereau J. Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J Exp Med. 2005;201(9):1479–86.
109 Allantaz F, Chaussabel D, Stichweh D, Bennett L, Allman W, Mejias A, et al. Blood leukocyte microarrays to diagnose systemic onset juvenile idiopathic arthritis and follow the response to IL-1 blockade. J Exp Med. 2007;204(9):2131–44.
110 Gattorno M, Piccini A, Lasiglie D, Tassi S, Brisca G, Carta S, et al. The pattern of response to anti-interleukin-1 treatment distinguishes two subsets of patients with systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2008;58(5):1505–15.
111 Picco P, Brisca G, Traverso F, Loy A, Gattorno M, Martini A. Successful treatment of idiopathic recurrent pericarditis in children with interleukin-1beta receptor antagonist (anakinra): An unrecognized autoinflammatory disease? Arthritis Rheum. 2009;60(1):264–8.
112 Schroder K, Zhou R, Tschopp J. The NLRP3 inflammasome: a sensor for metabolic danger? Science. 2010;327(5963):296–300.
113 Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;466(7306):652.
Funding / potential competing interests: No financial support and no other potential conflict of interest relevant to this article was reported.