Inhaled nanoparticles and lung cancer – what we can learn from conventional particle toxicology
DOI: https://doi.org/10.4414/smw.2012.13547
Ken
Donaldson, Craig A.
Poland
Summary
Manufactured nanoparticles (MNP) represent a growth area in industry where their interesting and useful properties bestow advantage over conventional particles for many purposes. This review specifically addresses the potential for lung cancer in those who might be exposed to airborne MNP. There is no strong evidence that MNP are carcinogenic and MNP come in a wide spectrum of materials, sizes, shapes and compositions and it is likely that the hazard will vary across different MNP types dependent upon their intrinsic properties. Low toxicity low solubility (LTLS) MNP are unlikely to pose a substantial cancer risk as they are not very biologically active. Nanoparticles with a more reactive surface may undoubtedly generate inflammation more readily and inflammation could be sufficiently intense to lead to secondary carcinogenesis via the oxidants and mitogens produced during inflammation. There is some evidence in vitro that MNP can gain access to the nucleus and the genetic material if specifically designed to do so by surface modification and that nanoparticles such as carbon nanotubes (CNT) can cause genetic aberrations by a primary mechanism additional to the inflammation–mediated one; these potential mechanisms require further study. High aspect ratio nanoparticles (HARN) are MNP that are fibre-shaped and analogously to asbestos might pose a special cancer hazard to the lungs, pleural and peritoneal mesothelium. Recent research suggests that the existing fibre pathogenicity paradigm is adequate for describing the hazard of HARN and that making the HARN of a non-biopersistent material or restricting the length could, via benign-by-design principles, allow safe HARN to be produced.
Background
Manufactured nanoparticles (MNP) are a diverse group of materials finding increasing use in manufacture, food and medicine due to their useful properties, but there has been concern over the risk to human health and the environment that these may pose [1, 2]. The potential adverse effects of MNP on human health have been discussed extensively but this short review specifically addresses the potential of inhaled MNP to cause lung cancer based on experience with other larger/conventional particles. There is little evidence at present showing that manufactured nanoparticles (MNP) cause cancer in humans. However, there may not have been sufficient duration of exposure to many of the newer forms of MNP for any effects to emerge, although bulk – manufactured nanoparticles (TiO2, carbon black, silica, alumina) have been produced for many decades with no evidence of an increased cancer risk from exposure to these particles in humans. However, it should be noted that the exposures in these industries must have been mixed exposures to nano- and micro- particles. In addition, in the eighties and nineties there were chronic inhalation studies with ultrafine (nanoparticulate) particles to address concerns over rat responses to low toxicity dusts such as TiO2 and carbon black. These studies revealed the phenomenon of ‘rat lung overload’ as a complicating factor in the carcinogenicity testing of low toxicity particles. There is also a large database of existing studies with conventional carcinogenic particles, such as quartz and asbestos and also studies with diesel exhaust nanoparticles as a workplace exposure. Inhalation studies with rats show that some particles are indeed capable of causing lung cancer in both humans and in animal models following long-term exposure and accumulation of sufficient dose. Here we address the question as to whether MNP have potential to cause cancer based on data from conventional particle toxicology and on data accumulated so far on MNP effects. The evidence to date does suggest that harmful nanoparticles have, in general, the same types of effects at the cellular level as harmful larger particles. It is important to draw a distinction between harmful and less harmful nanoparticles since there is ample evidence that nanoparticles, like larger particles, are heterogeneous in terms of the type and intensity of the hazard they pose following deposition in the lungs. Therefore, it is unwise to discuss nanoparticles as if they were a one hazard entity. The types of adverse effects that harmful particles and nanoparticles produced include the ability to cause oxidative stress, inflammation and genotoxicity, all of which are pathobiologically linked to cancer. Therefore, it may be argued that demonstrations that MNP in vitro are capable of having these effects raises concern that in the long-term, given sufficient exposure to some MNP and accumulation of dose in target tissues, cancer could be produced in some populations. This paper provides some of the background to the issue of the potential carcinogenicity of MNP based on the toxicological literature and especially seeks to use longer-term toxicological experience with conventional carcinogenic particles to contextualise the more limited existing data on MNP carcinogenesis.
Biological activities of MNP linked to cancer
As mentioned, there is little evidence currently as to the carcinogenic effects of MNP in humans, in addition there are few if any recent animal studies addressing a cancer endpoint for MNP. However, some useful information may be gained from the literature on the effects of MNP in vitro and in short term studies that shed light on a likely carcinogenic mechanism. We focus on pathobiological processes considered most relevant to particles carcinogenesis.
Oxidative stress
Oxidative stress is one of the most common endpoints reported following the treatment of cells in culture with MNP. We have previously pointed out that, so common is this endpoint in terms of its reporting that it does not add to the general sum of knowledge to describe any more instances where MNP cause oxidative stress [3]. That is not to say that oxidative stress is not important but the extent, type and cellular location needs to be understood rather than simply describing its existence in MNP-treated cells. For example the production of oxidative DNA adducts has more direct relevance to carcinogenesis than depletion of glutathione [4]. The relative importance of oxidative stress lies in the ability of oxidative stress to mediate a number of active processes in the cells, such as apoptosis, DNA adduct formation and pro-inflammatory gene expression. All of these have been reported following treatment with some types of MNP at various doses [4-8]. The severity of the oxidative stress may be an important step in triggering these processes in a tiered way [9]. Oxidative stress may be caused directly by particle structures generating ROS in the vicinity or inside the cell or could arise more indirectly due to the effects of internalised particles on mitochondrial respiration [10] or in depletion of antioxidant species within the cell [11].
Inflammation
There is a close connection between oxidative stress in the cell and the elicitation of an inflammatory response via pro-inflammatory gene transcription. In addition, there may be oxidation of lipid species within the cell that lead to the production of pro-inflammatory eicosanoids [12]. Many studies have reported pro-inflammatory effects of MNP at various doses on pro-inflammatory gene expression in the cell [8]. Pro-inflammatory pathways such as the MAP kinases are oxidative stress-responsive and so play a role in gene expression and are activated by some nanoparticles [13]. The redox-responsive NF-κB and AP-1 transcription factors have also reported to be activated in MNP exposed cells [14] [15]. In addition there are numerous studies reporting the induction of inflammation itself in the lungs following deposition of MNP by intratracheal installation [8] or inhalation exposures [16].
Genotoxicity
A substance is considered genotoxic if it deleteriously affects the genome of a cell either by direct or indirect damage to the cellular DNA including effects on the cellular pathways that monitor and protect genome integrity. In situations where a particle or substance directly interacts with the genomic DNA and causes damage, the effect is said to be direct primary genotoxicity and this, by its nature is independent of inflammation [17]. This can include the formation of DNA lesions such as 8-oxo-7,8-dihydro-2'-deoxyguanosine due to intrinsic free radical production from the particle or through direct binding of the particle with the DNA or component of the cell division machinery such as centromeres or microtubule spindle causing an aneugenic effect. Indirect primary genotoxicity in contrast occurs when intracellular antioxidants are depleted and therefore results in an imbalance between cellular steady-state oxidants (i.e. produced during normal cellular activities such as respiration) and the depleted anti-oxidants leading to a situation of oxidative stress driven genotoxicity [4].
Secondary genotoxicity does not involve the direct interaction of a particle with the target cell in which genotoxicity occurs but instead drives genotoxicity through its interaction with other cells causing the production of an environment conducive to the accumulation of genetic damage and proliferation. This is typically caused by the induction of chronic inflammation leading to persistent oxidative stress caused by the presence inflammatory cells such as macrophages and polymorphonuclear leukocytes as well as the secretion of various pro-survival and proliferation signalling factors.
In reality the induction of genotoxic effects can be due either to direct or indirect primary genotoxicity, secondary genotoxicity or a mixture of all as the production of reactive species within in a cell is also likely to drive inflammatory signalling through activation of oxidant-sensitive transcription factors such as NF-κB and AP-1 [18].
Fibrosis
In a number of studies fibrosis has been described as an endpoint following MNP deposition in the lungs. These effects appear to be driven by conventional inflammatory effects, but also by unusual modes of inflammation including eosinophils [8] and also, in the case of carbon nanotubes, a propensity for fibroblasts to grow along interstitialised carbon nanotubes [19]. There is a well-documented link between fibrosis in interstitial tissue and transformation in the associated epithelium in a number of lung conditions, such as idiopathic pulmonary fibrosis, asbestosis and silicosis [20–22]. Based on this link, one could consider the possibility that any MNP that were to induce fibrosis to a substantial degree could therefore have the potential to cause lung cancer. However, as in all cases, the carcinogenic endpoint is dose-related and so the issue of exposure is indeed critical and needs to be taken into consideration. Toxicological studies are concerned with hazard and determining the threshold at which adverse effects occur and as such the doses used are often very high. Risk assessment seeks to contextualise the doses that demonstrate a hazard using dosimetric and cross-species considerations to decide whether at plausible exposures these doses would be attained and whether the model used is a relevant one for predicting human hazard.
Conventional particles and cancer
Quartz
Quartz (or crystalline silica) exposure is associated with silicosis and also with the development of lung cancer [22]. In 1996 a working group of the World Health Organisation (WHO) international agency for research on cancer (IARC) reviewed the published literature on quartz and lung cancer. They concluded that there was “sufficient evidence in humans for the carcinogenicity of inhaled crystalline silica in the form of quartz or crystalline silica from occupational sources” [22]. Animal and experimental cell data were also taken into consideration and there was firm supporting evidence that quartz was capable of producing genotoxicity, inflammation, and oxidative stress that could contribute to carcinogenesis. The mechanism of quartz genotoxicity can be primary, as a result of direct interaction of quartz with target cells, or indirect as a consequence of inflammation elicited by quartz [22]. In the latter case, inflammatory cell derived oxidants are considered to cause the genotoxicity. A recent review by one of the authors [23] covered the intervening literature between the reclassification in 1997 and the present date to determine the state of the science on the mechanism of quartz carcinogenesis. The data strongly suggested that inflammation was the driving force for genotoxicity and that primary genotoxicity of deposited quartz would play a role only at very high, possibly implausible, exposures and deposited doses [23]. This is supported by a recent re-evaluation of silica by IARC where the established mechanistic events given are ‘Impaired particle clearance leading to macrophage activation and persistent inflammation’ with no mention of primary genotoxicity [24].
Asbestos
In addition to causing a number of pleural and lung parenchymal conditions, the cancers associated with exposure to asbestos are lung cancer (bronchogenic carcinoma) and mesothelioma. Asbestos minerals are crystalline with weaknesses in the crystal structure which cause long thin fibres to be released along fracture planes and become airborne when the rock is stressed. These high aspect ratio (length to width) fibres are thin enough to be inhaled into the lung where they are variably pathogenic, depending on the type of asbestos. This is due to the compliance of asbestos with a fibre pathogenicity paradigm by which the properties of fibres are understood to affect pathogenicity [25]. The important factors are diameter, length and biopersistence and long (longer than ~10 µm), thin and biopersistent fibres are capable of causing a number of processes in the lung that could lead to carcinogenesis. The lack of biopersistence of chrysotile causing it to disintegrate and become shorter in the lungs is the main structural feature that leads to chrysotile being less pathogenic than the amphiboles [26]. Long biopersistent fibres can chronically generate free radicals that directly damage DNA due to difficulties encountered in clearing such fibres, leading to long residence time, and accumulation of dose, as well as interactions with cells of the immune system. In addition, iron which is integrated into the crystalline structure of asbestos can contribute to redox-cycling reactions leading to the production of damaging hydroxyl radicals [27]. Fibres may also physically interfere with mitosis of cells in culture, a mechanism whereby asbestos and other types of fibres are able to induce aneuploidy and polyploidy [28]. The mechanism envisaged here is that once inside the cell, the long fibres may physically interfere with mitosis by interacting with the mitotic spindle and chromosome segregation resulting in slowly moving or sticky chromosomes during anaphase leading to production of aneuploid daughter cells [28]. Long fibres are also capable of stimulating proliferation which generally adds to the likelihood of any mutations or carcinogenic effects being fixed in [29]. Long thin fibres can also provoke a chronic inflammatory response via frustrated phagocytosis [25] where macrophages that are unable to fully enclose a long fibre are chronically stimulated to release cytokines and other histotoxic products.
Diesel exhaust particulate
Whilst there is some debate in the published literature there is persuasive evidence linking diesel exhaust particulate (DEP) exposure to lung cancer [30–32]. Whilst diesel exhaust particulate is a nanoparticle, it is significantly different from the MNP by the fact that DEP are very complex and contain a mixture of combustion-derived chemicals, many of which could be carcinogenic i.e. metals and a wide variety of organics [33]. Subsequently the underlying mechanism for DEP-induced lung cancer has been a matter of considerable argument. Central to this question has been whether adsorbed mutagenic organic compounds such as polycyclic aromatic hydrocarbons (PAH) are involved or whether the carbon core itself represents the biologically effective dose (i.e., the entity which drives the effect). In studies in rats comparing high exposure to carbon black which contains very little absorbed organic compounds, with diesel particles which contain high amounts of organic compounds, there was little difference in the tumour response or the genetic defects found in the tumour cells [34, 35]. Neither was there very much in the way of additional bulky DNA adducts found in the diesel soot exposed animals, which would have been produced by the organic fraction on the DEP whilst both diesel and carbon black produced approximately the same amount of lung inflammation and tumour response [36]. It was concluded therefore that the carbonaceous core, by virtue of its ability to cause inflammation and proliferation, was the predominant factor involved in diesel soot carcinogenesis. It’s not known whether rats are uniquely different from humans and whether the mechanism is different in humans. Epigenetic mechanisms play a role in carcinogenesis and cellular functions, including the regulation of inflammatory gene expression, DNA repair, and cell proliferation, can be regulated by epigenetic changes such as DNA methylation. An environmental exposure like diesel exhaust has potential to induce an epigenetic change and more research is needed in this area [37].
Conclusions regarding mechanism
In general, these data from the different particulate carcinogens show and imply a rather similar pattern of underlying processes, namely oxidative stress, inflammation, proliferation, mutation etc leading to tumour response. The clear implication is that in relation to particles, the inflammation is the primary driver of the carcinogenesis. In support of this concept, Driscoll et al. [38] showed HPRT-mutations in alveolar epithelial cells isolated and cultured from rats that had been exposed to carbon black, titanium dioxide or quartz. Importantly only the exposures causing a significant persistent inflammation in the rat lungs induced mutations [39]. In parallel experiments, respirable quartz particles were unable to cause increased HPRT mutations in rat lung epithelial cells in vitro, whereas lavaged inflammatory cells (macrophages and neutrophils) from quartz-exposed rat lungs were capable of causing the same mutations in vitro [39]. The importance of inflammation emphasises the need to understand how particle properties might lead to inflammation. In a number of studies the surface area of particles was found to drive overload-type inflammation [40] and carcinogenesis in rats [41]. Nanoparticles have a high surface area per unit mass and so any biological effect that is driven by surface area can be driven by a low mass (high surface area) dose of nanoparticles [42]. The initiation of inflammation by LTLS particles in rats is part of a process known as ‘rat lung overload’. This condition is confined to rats and is not found in other rodents or in humans and arises at high exposure concentrations or lung burdens to LTLS dusts where there is inflammation and a build-up of dose due to impaired clearance [43]; it has no meaning for dusts that are intrinsically toxic and can only be applied to LTLS dusts. In chronic inhalation studies in rats with these particles, pulmonary fibrosis and lung tumours were present at high exposure concentrations where there was a build-up of very high lung burdens. Rat lung overload and the attainment of such high lung burdens is a consequence of a sequence of processes roughly in the following order – failed clearance with accumulation of dose → inflammation → altered particle kinetics with retention consequent on impaired clearance → fibrosis → proliferation → benign and malignant lung tumours [44]. This process and tumours that it produces are considered to have little relevance for human risk assessment since they are confined to the rat and its unique response to high levels of low toxicity surface in the lungs. However, surface area dose can lead to inflammatory responses in non-overload conditions i.e. at low surface area burdens [42, 45].
Manufactured nanoparticles
When considering the potential genotoxicity and carcinogenicity of MNP, one has to consider if MNP are likely to behave in a similar manner to conventional particles as described above or if there are potentially new routes of genotoxicity which may drive a carcinogenic effect. Given their small size, an area for concern maybe the ability of nanoparticles to penetrate cells and accumulate within hitherto privileged cellular compartments such as mitochondria or nucleus where their presence may have deleterious effects. This could be considered especially relevant for nanoparticles specifically designed to penetrate cellular membranes and target structures such as the nucleus for the purpose of delivering some form of therapeutic payload.
In the 2008 study by Nativo et al, the authors tried successfully to circumvent the endosomal uptake of 16nm gold nanoparticles to deliver the particles into the cytosol of the cell [46]. This was done using either encapsulation of the particles within lipid vesicles which, when fused with the cell membrane delivered the particles to the cytosol, or via the surface modification of the gold nanoparticles with ‘cell-penetrating peptides’ (CPPs) [47]. Using a combination of oligopeptides, the authors demonstrated that they could achieve particles freely dispersed in the cytosol and with the addition of a nuclear localisation sequence on the surface of the particles, could also target the particles to the nucleus where interaction with DNA would be possible. It has also been shown using negatively charged carboxylated polystyrene beads ranging in size from 30 nm to 500 nm that the entry pathways of the beads moved from endocytosis to direct penetration (and associated sub-cellular localisation) with decreasing size [48]. However, if size can have such a profound effect on the route of cellular penetration and subcellular localisation, it is therefore also likely to be dependent on the agglomeration state of the nanoparticles which can be highly dynamic.
One of the confounding issues of trying to understand the carcinogenicity of particles is the potential for them to generate false-positive or false-negative results. Indeed it has been noted assays such as the Ames test may not be suitable for use with particulates including nanoparticles as the bacterial cell wall may act as a barrier [7] leading to the production of false-negative results (except perhaps in situations where it is a soluble component released from the particle which could drive genotoxicity).
As already mentioned, rat lung overload is a phenomenon associated with the overloading of lung clearance mechanisms leading to a rapid accumulation of particles in the lung leading to chronic inflammation. This in turn is likely to generate fibroproliferative changes including alveolar cell proliferation (hyperplasia), the conversion of cells to cell types not normally associated with the specific lung location (metaplasia). The overall consequence of lung overload may therefore involve local tumour formation (neoplasia) [44, 49, 50]. As we know, rats are particularly susceptible to lung overload during chronic exposure to high dose of LSLT particles and indeed in a study looking at the respiratory effects of nano-particulate TiO2, inhalation in 3 species (rats, mice and hamsters) noted that the initial lung burden of particles was approximately 77% lower in hamsters than rats and mice which they attributed to efficient lung clearance [51]. Indeed they saw significant induction of inflammation at the highest dose which was most severe in rats, with only mild inflammation seen in mice and no inflammation noted in hamsters at the same dose and post-exposure time point. The potential driver of the effect behind rat lung overload is particularly relevant to nanomaterials and could be seen as a confounding factor casting doubt on the validity of carcinogenic results in a rat lung model using high dose LSLT particles. It has been suggested that the driving metric (the biologically effective dose) is particle surface area and there have been several studies which show that particle surface area correlates well with induced pathogenic events after rat lung inhalation [52, 53]. One of the most convincing examples was a retrospective study by Tran et al. [40] in which initiation of inflammation following inhalation exposure to two LSLT (TiO2 and barium sulphate) was related to several dose metrics. Tran and colleagues found that if lung burden was expressed in terms of particle surface area there was a clear relationship with the level of inflammation and translocation to the lymph nodes, a marker of overload. From this relationship, the authors proposed that a threshold dose of approximately 200–300 cm2 led to lung overload in the rat model. Based on this metric and larger surface area to volume/mass ratio which typifies nanoparticles, it is clear to see how the potential for overload effects may be increased with those nanomaterials which exhibit a high biologically-accessible surface area. Other metrics have been suggested within the literature including particle mass and particle volume. Morrow [54] hypothesised that in rats, a reduction in macrophage mobility (and hence clearance rate) begins when 6 percent of the macrophage volume is filled with particles. Once macrophage volume reaches approximately 60% there is a total cessation of Alveolar macrophage-mediated clearance and this driver of lung overload has also been more recently suggested for carbon nanotubes which form low density wool-like clumped structures [55].
Despite this apparent ability of nanoparticles to achieve an overload threshold in rats at a lower mass dose than large, fine particles, the question still remains if carcinogenicity results from studies with rats using high dose repeated exposure to LSLT particles are relevant to human risk assessment due to the suggested differences in species susceptibility introducing further uncertainty. This is important when considering the potential for false-positive results gained from species-specific effects and in particular the difficulties faced in basing risk assessment for particle carcinogenicity on high exposure/dose studies in rats. The existence of the issue of particle overload emphasies the importance of low dose, repeat studies which is more closely associated with actual workplace exposure to particles in determining potential pathogenicity whilst avoiding potentially irrelevant and confounding effects.
This issue of high dose exposure leading potentially to misleading outcomes is not specific to the issue of inhalation and lung overload. It is also a common occurrence when looking at other systems and particularly in relation to in vitro toxicology where it can be difficult to relate the dose within a cell culture dish to that which would plausibly be encountered if that cell was within its natural environment such as the alveolar epithelium of the lung. Therefore, it is critical to consider dose both in terms of total dose applied and the dosing regime (single vs. repeat lower dose exposure) and the mechanism of effect before potential toxic outcomes such as carcinogenicity are extrapolated to a human system. Indeed even the exposure system can have profound effects on the observed outcomes which need to be taken into account when considering the likelihood of such effects occurring in humans exposed either occupationally or via the environment. For example, in one study [16] the authors observed mutations within the k-ras oncogene within the lungs of mice after exposure to SWCNT but most notably they found a greater frequency after inhalation of the SWCNT than instillation (a commonly used surrogate for inhalation exposure) which is likely related to the increased level of Inflammation, oxidative stress and fibrosis noted after inhalation [16].
Potential mechanisms of MNP-mediated lung cancer
In this section the various mechanisms by which MNP might have carcinogenic effects are discussed by analogy with existing particulate carcinogens for which more information is available.
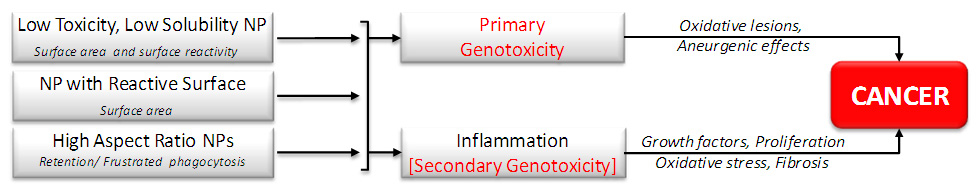
Figure 1
Diagram showing the potential processes that might be involved in MNP carcinogenesis. Biologically effective doses are shown below each particle type on the left and potentially important pathobiological processes are shown around the arrows leading to cancer on the right.
Analogous to quartz
The defining feature of quartz in particle toxicology terms and its ability to have a biological impact is its surface reactivity. The quartz surface is highly charged and interacts with membranes and proteins leading to its oxidative stressing and inflammogenic effects and these are directly related to the ability of quartz to cause genotoxicity and carcinogenesis [56]. MNP with a reactive surface (e.g., NiO2nanoparticles), are most likely to behave like quartz via the above mechanisms. We have for example described a panel of metal oxide MNP with varying surface charge, which is one measure of surface reactivity. Those materials with a high positive zeta potential were haemolytic, as is quartz [57] by virtue of its direct surface reactive effect on erythrocyte membranes. They were also inflammogenic, like quartz, but at a given mass has much more surface area than quartz and so are potentially more inflammogenic per unit mass than quartz [42]. For these reasons it would seem that reactive MNP would be most likely to act like quartz and may be able to cause inflammation and so could cause lung cancer as a consequence of inflammation–mediated carcinogenesis, given sufficient prolonged exposure.
Analogous to asbestos
High aspect ratio nanomaterials (HARN) have come under suspicion due to their similarity to asbestos [25] which can cause lung cancer and mesothelioma. The fibre pathogenicity paradigm (mentioned above) describes thinness, length and biopersistence as being the factors that determine whether a fibre is pathogenic or not for lung cancer and mesothelioma. HARN represent a growth area in nanotechnology because of the useful properties of fibres in terms of lightness, insulation and electrical properties [58]. Carbon nanotubes are the best known and studied of the HARN and are synthesised and handled in large quantities [58] with subsequent potential for human inhalation exposure. Other types of HARN are increasingly being developed for a range of uses. These nanorods, nanowires and nanofibres warrant close scrutiny as to whether they will produce effects like asbestos. In this regard the authors have set out to determine whether HARN conform to the fibre pathogenicity paradigm and initial studies demonstrate that HARN such as carbon nanotubes and nickel oxide nanowires show length-dependent mesothelial pathogenicity, similar to asbestos [59–61]. These studies do not extend to cancer endpoints but we suggest that short-term inflammogenicity and fibrogenicity may be mechanistically or predictively linked to long-term carcinogenicity. Furthermore, carbon nanotubes have the potential to be biopersistent as judged by in vitro durability studies at acid pH [62]. However, it should be noted that under conditions where the carbon nanotubes have been subjected to oxidation following strong acid treatment, they may be sufficiently fragile to undergo breakage and shortening under oxidative stress conditions such as would occur in lung [63]. The predictive strength of the fibre pathogenicity paradigm suggests that any HARN that is long, thin and biopersistent has the potential to be carcinogenic at the pleura and in the lung leading to mesothelioma and lung cancer if the exposure was great enough for the dose to accumulate sufficiently to initiate these processes.
As well as the evidence suggesting that HARN can drive frustrated phagocytosis leading to inflammation and oxidative stress in the surrounding tissue, there is also evidence analogous to asbestos that CNT can interfere with normal mitosis leading to the induction of aneuploidy and polyploidy. Treatment of cultured primary and immortalised human airway epithelial cells with single walled carbon nanotubes for 24hrs led to a variety of abnormalities including fragmented centrosomes, multiple mitotic spindle poles, anaphase bridges, and aneuploid chromosome number [64]. Most strikingly the authors of this study showed nanotubes within the nucleus of the treated cells and these were associated with cellular and mitotic tubulin. Whilst the exact mechanism driving the observed abnormalities are unknown, what is clear is that there is penetration of the cell with these fibrous nanomaterials and interaction with the cellular DNA, centrosomes and spindle apparatus which is associated with the formation of defects in cell division. The authors also noted that the diameter of the SWCNT bundles were comparable to that of the cellular and mitotic microtubules (~25 nm) [65] and that potentially this similarity in structure may have allowed the incorporation of internalised SWCNT into the mitotic spindle leading to abnormal cell division [65].
Analogous LTLS nanoparticles
There is a problem in interpreting lung tumour response seen in rats to LTLS particles and DEP since there are questions as to whether the rat serves as a useful model for human cancer resulting from these materials [66]. This is not the case for the other materials described here since the rat response to quartz and asbestos fibres fairly well reflects the mechanism and types of cancer seen in human populations exposed to these dusts. The complicating factor for LTLS particles is rat lung overload as described above. Overload tumours do not arise in humans and so the production of cancer in rats following overload is difficult to interpret. As has been shown for carbon black, titanium dioxide and other ultrafine or nanoparticulate LTLS particles, rats do show a tumour response to high lung burdens of this material. However, the lung burdens are very large and not plausibly produced by the exposures likely to arise in modern hygiene–regulated workplaces. There is no evidence of lung tumour response in humans exposed to high levels of LTLS dusts and so it seems on the face of it that a tumour response in humans at plausible exposures to LTLS nanoparticles is unlikely. The highest known exposures of any population to low toxicity particles are coal miners and there is no evidence of any tumour response in coal miners, despite massive lung burdens in terms of particle mass and surface area, of coal mine dust. That is not to say that there is not likely to be some kind of pathology produced by LTLS nanoparticles in the lungs if high lung burdens were to be produced but it is unlikely that a tumour response would be produced unless the response is fundamentally different to that seen with other LTLS nanoparticles. In that case the particles would not come under the heading of LTLS but might come under the heading of ‘analogous to quartz’.
Conclusion
In conclusion it is important to note that there is as yet no conclusive evidence that MNP are carcinogenic. However, based upon the what we understand from conventional particle toxicology and links between important aspects such as intrinsic reactivity and processes such as inflammation and the causation of carcinogenic changes, there is indeed the potential for some MNP to be carcinogenic given sufficient high and extended period of exposure. LTLS MNP are unlikely to pose much of a cancer risk as they are not very biologically active. Lifetime inhalation carcinogenesis studies in the nineties with carbon black, TiO2 and diesel soot nanoparticles demonstrated that cancer only occurred at low level and at extremely high lung burdens arising from very intense lifelong exposures as a consequence of ‘rat lung overload’ , which does not occur in humans.
Importantly, such cancers as those which did occur were confined to the lungs and so there is no reason to think that translocation away from the lungs to secondary target organs (except for the peritoneal cavity with HARN analogous to peritoneal mesothelioma arising in asbestos-exposed workers) might lead to cancer at any other sites. As described, there is some evidence in vitro that MNP can gain access to the nucleus and the genetic material especially if specifically designed to do so and that nanoparticles such as CNT can cause genetic aberrations by a primary mechanism additional to the inflammation–mediated one which requires further study. Nanoparticles with a more reactive surface may undoubtedly generate inflammation more readily. If the exposure and subsequent dose was sufficiently high and the inflammation sufficiently intense and chronic, reactive MNP could more readily lead to secondary carcinogenesis via inflammation. HARN pose a more complex and different hazard because they may be, analogously to asbestos, carcinogenic to the lungs, pleura and peritoneal cavity as a consequence of their fibrous structure. Recent research by the authors suggests that the existing fibre pathogenicity paradigm is adequate for describing the hazard of HARN exposure and that making the HARN of a non-biopersistent material or restricting the length could, via benign-by-design principles, allows safe HARN to be produced [58]. In all of the above it is important to point out that, analogously to conventional particle-induced cancer, these processes leading to cancer would only be initiated by protracted exposure to high airborne levels of MNP culminating in a high dose to the lungs.
References
1 The Royal Society and the Royal Academy of Engineering: Nanoscience and nanotechnologies: opportunities and uncertainties. R 2004.
2 Maynard AD, Aitken RJ, Butz T, Colvin V, Donaldson K, Oberdorster G, et al. Safe handling of nanotechnology. Nature. 2006;444:267–9.
3 Donaldson K, Borm PJ, Castranova V, Gulumian M. The limits of testing particle-mediated oxidative stress in vitro in predicting diverse pathologies; relevance for testing of nanoparticles. Part Fibre Toxicol. 2009;6:13.
4 Donaldson K, Poland CA, Schins RP. Possible genotoxic mechanisms of nanoparticles: criteria for improved test strategies. Nanotoxicology. 2010;4:414–20.
5 Moller P, Jacobsen NR, Folkmann JK, Danielsen PH, Mikkelsen L, Hemmingsen JG, et al. Role of oxidative damage in toxicity of particulates. Free Radic Res. 2010;44:1–46.
6 Li N, Xia T, Nel AE. The role of oxidative stress in ambient particulate matter-induced lung diseases and its implications in the toxicity of engineered nanoparticles. Free Radic Biol Med. 2008;44:1689–99.
7 Landsiedel R, Kapp MD, Schulz M, Wiench K, Oesch F. Genotoxicity investigations on nanomaterials: methods, preparation and characterization of test material, potential artifacts and limitations – many questions, some answers. Mutat Res. 2009;681:241–58.
8 Cho WS, Duffin R, Poland CA, Duschl A, Oostingh GJ, MacNee W, et al. Differential pro-inflammatory effects of metal oxide nanoparticles and their soluble ions in vitro and in vivo; zinc and copper nanoparticles, but not their ions, recruit eosinophils to the lungs. Nanotoxicology. 2011.
9 Nel A, Xia T, Madler L, Li N. Toxic potential of materials at the nanolevel. Science. 2006;311:622–7.
10 Xia T, Kovochich M, Liong M, Zink JI, Nel AE. Cationic polystyrene nanosphere toxicity depends on cell-specific endocytic and mitochondrial injury pathways. ACS Nano. 2008;2:85–96.
11 Park EJ, Yi J, Chung KH, Ryu DY, Choi J, Park K. Oxidative stress and apoptosis induced by titanium dioxide nanoparticles in cultured BEAS-2B cells. Toxicol Lett. 2008;180:222–9.
12 Liu X, Whitefield PD, Ma Y. Quantification of F(2)-isoprostane isomers in cultured human lung epithelial cells after silica oxide and metal oxide nanoparticle treatment by liquid chromatography/tandem mass spectrometry. Talanta. 2010;81:1599–606.
13 Lee HM, Shin DM, Song HM, Yuk JM, Lee ZW, Lee SH, et al. Nanoparticles up-regulate tumor necrosis factor-alpha and CXCL8 via reactive oxygen species and mitogen-activated protein kinase activation. Toxicol Appl Pharmacol. 2009;238:160–9.
14 Mroz RM, Schins RP, Li H, Drost EM, MacNee W, Donaldson K. Nanoparticle carbon black driven DNA damage induces growth arrest and AP-1 and NFkappaB DNA binding in lung epithelial A549 cell line. J Physiol Pharmacol. 2007;58(Suppl 5):461–70.
15 Li R, Ning Z, Majumdar R, Cui J, Takabe W, Jen N, et al. Ultrafine particles from diesel vehicle emissions at different driving cycles induce differential vascular pro-inflammatory responses: implication of chemical components and NF-kappaB signaling. Part Fibre Toxicol. 2010;7:6.
16 Shvedova AA, Kisin ER, Murray AR, Johnson VJ, Gorelik O, Arepalli S, et al. Inhalalation versus aspiration of single walled carbon nanotubes in C57/Bl6 mice: Inflammation, fibrosis, oxidative stress and mutagenesis. Am J Physiol Lung Cell Mol Physiol. 2008;295:L552–L565.
17 Schins RP, Knaapen AM. Genotoxicity of poorly soluble particles. Inhal Toxicol. 2007;19(Suppl 1):189–98.
18 Sen CK, Packer L. Antioxidant and redox regulation of gene transcription. FASEB J. 1996;10:709–20.
19 Wang L, Mercer RR, Rojanasakul Y, Qiu A, Lu Y, Scabilloni JF, et al. Direct fibrogenic effects of dispersed single-walled carbon nanotubes on human lung fibroblasts. J Toxicol Environ Health A. 2010;73:410–22.
20 Ardies CM. Inflammation as cause for scar cancers of the lung. Integr Cancer Ther. 2003;2:238–46.
21 Kamp DW. Asbestos-induced lung diseases: an update. Transl Res. 2009;153:143–52.
22 IARC. IARC monographs on the evaluation of carcinogenic risks to humans. Volume 68. Silica, some silicates, coal dust and para-aramid fibrils. 1997. Ref Type: Generic
23 Borm PJA, Donaldson K. The carcinogenic action of crystalline silica (CS): a review of the evidence supporting indirect, inflammation-driven genotoxicity as a principal mechanism. CRC Critical Reviews in Toxicology (in press). 2011. Ref Type: Generic
24 Straif K, Benbrahim-Tallaa L, Baan R, Grosse Y, Secretan B, El Ghissassi F, et al. A review of human carcinogens-part C: metals, arsenic, dusts, and fibres. Lancet Oncol. 2009;10:453–4.
25 Donaldson K, Murphy FA, Duffin R, Poland CA. Asbestos, carbon nanotubes and the pleural mesothelium: a review of the hypothesis regarding the role of long fibre retention in the parietal pleura, inflammation and mesothelioma. Part Fibre Toxicol. 2010;7:5.
26 Hodgson JT, Darnton A. The quantitative risks of mesothelioma and lung cancer in relation to asbestos exposure 7. Ann Occup Hyg. 2000;44:565–601.
27 Lund LG, Aust AE. Iron mobilization from crocidolite asbestos greatly enhances crocidolite-dependent formation of DNA single-strand breaks in phi X174 RFI DNA. Carcinogenesis. 1992;13:637–42.
28 Kane AB. Mechanisms of mineral fibre carcinogenesis. In: Mechanisms of fibre carcinogenesis. Edited by Kane AB, Boffetta P, Saracci R and Wilbourn JD 1996, 11–34.
29 Brody AR, Overby LH, Warheit DB. Inhaled asbestos attracts macrophages and induces rapid proliferation of bronchiolar-alveolar cells. Chest. 1987;91:302.
30 Ris C. U.S. EPA health assessment for diesel engine exhaust: a review. Inhal Toxicol. 2007;19(Suppl 1):229–39.
31 Olsson AC, Gustavsson P, Kromhout H, Peters S, Vermeulen R, Bruske I, et al. Exposure to diesel motor exhaust and lung cancer risk in a pooled analysis from case-control studies in Europe and Canada. Am J Respir Crit Care Med. 2011;183:941–8.
32 Lipsett M, Campleman S. Occupational exposure to diesel exhaust and lung cancer: a meta-analysis. Am J Public Health. 1999;89:1009–17.
33 Wichmann HE. Diesel exhaust particles. Inhal Toxicol. 2007;19(Suppl 1):241–4.
34 Nikula KJ, Snipes MB, Barr EB, Griffith WC, Henderson RF, Mauderly JL. Comparative pulmonary toxicities and carcinogenicities of chronically inhaled diesel exhaust and carbon-black in f344 rats. Fundamental And Applied Toxicology. 1995;25:80–94.
35 Heinrich U, Fuhst R, Rittinghausen S, Creutzenberg O, Bellmann B, Koch W, et al. Chronic inhalation exposure of wistar rats and 2 different strains of mice to diesel-engine exhaust, carbon-black, and titanium-dioxide. Inhalation Toxicology. 1995;7:533–56.
36 Gallagher J, Heinrich U, George M, Hendee L, Phillips DH, Lewtas J. Formation of dna-adducts in rat lung following chronic inhalation of diesel emissions, carbon-black and titanium-dioxide particles. Carcinogenesis. 1994;15:1291–9.
37 Bollati V, Baccarelli A. Environmental epigenetics. Heredity. (Edinb ) 2010;105:105–12.
38 Driscoll KE, Carter JM, Howard BW, Hassenbein DG, Pepelko W, Baggs RB, et al. Pulmonary inflammatory, chemokine, and mutagenic responses in rats after subchronic inhalation of carbon-black. Toxicology And Applied Pharmacology. 1996, 136.
39 Driscoll KE, Deyo LC, Carter JM, Howard BW, Hassenbein DG, Bertram TA. Effects of particle exposure and particle-elicited inflammatory cells on mutation in rat alveolar epithelial cells. Carcinogenesis. 1997;18:423–30.
40 Tran CL, Buchanan D, Cullen RT, Searl A, Jones AD, Donaldson K. Inhalation of poorly soluble particles. II. Influence of particle surface area on inflammation and clearance 1. Inhal Toxicol. 2000;12:1113–26.
41 Driscoll KE. Role of inflammation in the development of rat lung tumors in response to chronic particle exposure. Inhalation Toxicology. 1996;8(suppl):139–53.
42 Duffin R, Tran L, Brown D, Stone V, Donaldson K. Proinflammogenic effects of low-toxicity and metal nanoparticles in vivo and in vitro: highlighting the role of particle surface area and surface reactivity 2. Inhal Toxicol. 2007;19:849–56.
43 Mauderly JL. Relevance of particle-induced rat lung tumors for assessing lung carcinogenic hazard and human lung cancer risk. Environmental Health Perspectives. 1997;105:1337–46.
44 Oberdorster G. Significance of particle parameters in the evaluation of exposure-dose-response relationships of inhaled particles. Particulate Science And Technology. 1996;14.
45 Gilmour PS, Ziesenis A, Morrison ER, Vickers MA, Drost EM, Ford I, et al. Pulmonary and systemic effects of short-term inhalation exposure to ultrafine carbon black particles. Toxicol Appl Pharmacol. 2004;195:35–44.
46 Nativo P, Prior IA, Brust M. Uptake and intracellular fate of surface-modified gold nanoparticles. ACS Nano. 2008;2:1639–44.
47 Bolhassani A. Potential efficacy of cell-penetrating peptides for nucleic acid and drug delivery in cancer. Biochim Biophys Acta. 2011;1816:232–46.
48 Zhang M, Li J, Xing G, He R, Li W, Song Y, et al. Variation in the internalization of differently sized nanoparticles induces different DNA-damaging effects on a macrophage cell line. Arch Toxicol. 2011.
49 Mauderly JL. Usefulness of animal-models for predicting human responses to long- term inhalation of particles. Chest. 1996;109: S.
50 Miller FJ. Dosimetry of particles in laboratory animals and humans in relationship to issues surrounding lung overload and human health risk assessment: a critical review. Inhal Toxicol. 2000;12:19–57.
51 Bermudez E, Mangum JB, Wong BA, Asgharian B, Hext PM, Warheit DB, et al. Pulmonary responses of mice, rats, and hamsters to subchronic inhalation of ultrafine titanium dioxide particles. Toxicol Sci. 2004;77:347–57.
52 Elder A, Gelein R, Finkelstein JN, Driscoll KE, Harkema J, Oberdorster G. Effects of subchronically inhaled carbon black in three species. I. Retention kinetics, lung inflammation, and histopathology. Toxicol Sci. 2005;88:614–29.
53 Borm PJ, Schins RP, Albrecht C. Inhaled particles and lung cancer, part B: paradigms and risk assessment. Int J Cancer. 2004;110:3–14.
54 Morrow PE. Possible mechanisms to explain dust overloading of the lungs. Fundam Appl Toxicol. 1988;10:369–84.
55 Pauluhn J. Subchronic 13-week inhalation exposure of rats to multiwalled carbon nanotubes: toxic effects are determined by density of agglomerate structures, not fibrillar structures. Toxicol Sci. 2010;113:226–42.
56 Borm PJ, Tran L, Donaldson K. The carcinogenic action of crystalline silica: a review of the evidence supporting secondary inflammation-driven genotoxicity as a principal mechanism. Crit Rev Toxicol. 2011;41:756–70.
57 Clouter A, Brown D, Hohr D, Borm P, Donaldson K. Inflammatory effects of respirable quartz collected in workplaces versus standard DQ12 quartz: particle surface correlates. Toxicol Sci. 2001;63:90–8.
58 Donaldson K, Murphy F, Schinwald A, Duffin R, Poland CA. Identifying the pulmonary hazard of high aspect ratio nanoparticles to enable their safety-by-design. Nanomedicine. (Lond) 2011;6:143–56.
59 Poland CA, Duffin R, Kinloch I, Maynard A, Wallace WA, Seaton A, et al. Carbon nanotubes introduced into the abdominal cavity of mice show asbestos-like pathogenicity in a pilot study. Nat Nanotechnol. 2008;3:423–8.
60 Murphy FA, Poland CA, Duffin R, Al Jamal KT, Ali-Boucetta H, Nunes A, et al. Length-dependent retention of carbon nanotubes in the pleural space of mice initiates sustained inflammation and progressive fibrosis on the parietal pleura. Am J Pathol. 2011;178:2587–600.
61 Poland CA, Byrne F, Cho WS, Prina-Mello A, Murphy FA, Davies GL, et al. Length-dependent pathogenic effects of nickel nanowires in the lungs and the peritoneal cavity. Nanotoxicology. 2011.
62 Osmond-McLeod MJ, Poland CA, Murphy F, Waddington L, MOrris H, Hawkins SC, et al. Durability and inflammogenic impact of carbon nanotubes compared with asbestos fibres. Part Fibre Toxicol. 2011;8:15.
63 Kagan VE, et al. Carbon nanotubes degraded by neutrophil myeloperoxidase induce less pulmonary inflammation. Nature Nanotechnology Advance online publication April 4th 2010. 2010. Ref Type: Generic
64 Sargent LM, Shvedova AA, Hubbs AF, Salisbury JL, Benkovic SA, Kashon ML, et al. Induction of aneuploidy by single-walled carbon nanotubes. Environ Mol Mutagen. 2009;50:708–17.
65 Sargent LM, Reynolds SH, Castranova V. Potential pulmonary effects of engineered carbon nanotubes: in vitro genotoxic effects. Nanotoxicology. 2010;4:396–408.
66 Mauderly JL. Lung overload:the dilemma and opportunities for resolution. Special issue. Particle overload in the rat lung and lung cancer:implications for risk assessment. Inhal Toxicol. 1996;8(Supplement):1–28. Ref Type: Generic