Interleukin-1, inflammasomes, autoinflammation and the skin
DOI: https://doi.org/10.4414/smw.2012.13590
Emmanuel
Contassot, Hans-Dietmar
Beer, Lars E.
French
Summary
Interleukin 1, one of the first cytokines discovered in the 1980s, and a potent mediator of fever, pain and inflammation, is at present experiencing a revival in biology and medicine. Whereas the mechanism of activation and secretion of interleukin 1β, which critically regulates the function of this molecule, has remained mysterious for some 30 years following its discovery, the identification of a new cytoplasmic complex of proteins regulating IL-1β activation and secretion has carried our understanding of the role of IL1 in biology and disease one big step further. The inflammasomes, recently identified innate immune complexes that sense intracellular danger- (e.g. uric acid, ATP, cytoplasmic DNA) or pathogen-associated molecular patterns (e.g. muramyl dipeptide, flagellin, anthrax lethal toxin), are now known to be responsible for triggering inflammation in response to several molecular patterns, including, for example, uric acid, a danger-associated molecular pattern and trigger of gout. Dysregulation of inflammasome function is however also the cause of a family of genetic autoinflammatory diseases known as cryopyrin-associated periodic syndromes (CAPS) characterised by recurrent episodes of fever, urticarial-like skin lesions, systemic inflammation and arthritis. In mouse models recapitulating mutations observed in CAPS, neutrophilic inflammation of the skin is a cardinal feature, in a manner similar to several autoinflammatory diseases with skin involvement such as PAPA (pyoderma gangrenosum, acne and pyogenic arthritis) and Schnitzler’s syndrome, in which IL-1β very probably plays a pathogenic role. In this article the role of the inflammasome in IL-1 biology, autoinflammation and disease is reviewed, together with new avenues for the therapy of these diseases.
Key words:
inflammation; interleukin-1; innate immunity; inflammasome; skin; fever
Introduction
In all types of tissue harmful stimuli such as trauma, pathogens or irritants induce a very complex response which is called inflammation [1]. This fundamental and beneficial process is required to remove the injurious stimulus and to initiate the healing process aiming to restore tissue homoeostasis. Although the quality and quantity of the inflammatory response depend on this stimulus and the context, the very initial steps of the inflammatory response are stereotyped as part of the innate immune response. Since antiquity it has been known that acute inflammation, which occurs within minutes or hours, is characterised by the cardinal signs redness, heat, pain, swelling and loss of function of the inflamed tissue [2]. However, inflammation can also become chronic and destructive, thus contributing to the pathogenesis of chronic inflammatory diseases. Recently evidence has accumulated showing that low-grade chronic inflammation can cause or contribute to the development of further diseases such as type 2 diabetes, Alzheimer`s disease, artherosclerosis, and cancer [1, 3].
The skin is our largest organ, which is exposed to and protects the body from microbes, pathogens and several irritants. In most cases, inflammation of the skin represents a protective process, e.g. upon injury or infection [4]. However, the skin can also be a site of excessive immune responses resulting in chronic inflammation, autoimmunity or autoiinflammation [5]. During the last ten years much progress has been made in our understanding of the molecular and cellular mechanisms underlying the initiation of an inflammatory response as part of the innate immune response, be it beneficial or unwanted. The cytokines that have proved essential at the onset of inflammation and during an innate immune response include the proinflammatory cytokine interleukin-1 (IL-1). This cytokine plays a fundamental role in regulating systemic, but also certain cutaneous inflammatory responses, and recently acquired knowledge regarding the processing and activation of IL-1 beta (IL-1β) has taught us that it is implicated in the pathogenesis of an emerging family of autoinflammatory diseases and fever syndromes [4, 6].
In this review we describe the key advances in the understanding of the biology and pathophysiological role of IL-1β with special focus on its contribution to autoinflammation and its function in the skin.
Interleukin-1 (IL-1)
IL-1 is an essential proinflammatory cytokine which possesses multiple properties and affects almost all cell types [6–7]. It is a mediator of the acute phase of inflammation by induction of local and systemic responses. Amongst others IL-1 induces the expression of adhesion molecules on endothelial cells, which are required for the infiltration of the stressed tissue by inflammatory and immunocompetent cells [6]. It also induces pain sensitivity, fever, vasodilation and hypotension. IL-1 is strongly expressed by monocytes, tissue macrophages and dendritic cells, but is also produced by B lymphocytes, NK cells and epithelial cells [4, 6]. Both IL-1α and IL-1β exert their activity by binding and signalling through IL-1 receptor type I (IL-1RI), which is expressed by almost all cell types. The cytoplasmic part of IL-1RI carries a toll/interleukin-1 receptor domain (TIR), which is also found in toll-like receptors (TLR), pointing to an important role of IL-1 in inflammation and innate immunity. Signal transduction by IL-1α and IL-1β requires recruitment of the IL-1R accessory protein (IL-1RAcP), and results in a series of incompletely understood phosphorylation and ubiquitination events, finally leading to activation of nuclear factor ĸB (NF-ĸB) and signalling by the c-Jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinase (MAPK) pathway [8]. IL-1R type II (IL-1RII) is a decoy receptor which lacks the TIR domain and cannot signal. This receptor is also released from cells and can thus also block the activity of IL-1 for neighbouring cells.
Agonists of IL-1RI are IL-1α and IL-1β, which are transcribed from separate genes. In contrast, binding of IL-R antagonist (IL-1Ra) to IL-1RI, which prevents binding of IL-1α and IL-1β, does not result in recruitment of IL-1RAcP and consequently leads to blockade of IL-1RI signalling [9]. Recombinant IL-1Ra (Kineret, Anakinra®) is used as a therapeutic in humans and has been approved for the treatment of rheumatoid arthritis [10]. In recent years it has emerged that Kineret shows clinical efficacy in the treatment of a relatively broad spectrum of inflammatory diseases without causing significant side effects, thus demonstrating the central role IL-1 plays in these conditions [7]. A disadvantage of Kineret is however its short half-life in vivo, which requires daily injection of the drug. This has been overcome by the recent development of additional inhibitors of IL-1 signalling, including a soluble receptor for IL-1 (Rilonacept, Arcalyst®), and a monoclonal antibody to IL-1β (Canakinumab, Ilaris®).
Regulation of IL-1 production
Since IL-1 is such a potent and important cytokine, it is not surprising that the generation of IL-1 activity is tightly regulated at different levels. Evidence suggests that the expression of IL-1α, although it can be regulated, exists to some extent at a constitutive level whereas in most cell types expression of IL-1β is only detectable upon stimulation [6, 11]. Expression of IL-1α and β is induced by activation of the transcription factor NF-ĸB, for example by exposure of macrophages and dendritic cells to the TLR4 agonist LPS (lipopolysaccharide) but also by IL-1 itself. In addition, it has been shown that IL-1β mRNA can be very unstable [12–13].
IL-1α and IL-1β are expressed as proproteins (proIL-1α and proIL-1β). ProIL-1β cannot bind to IL-1RI, since receptor binding and activation requires prior proteolytic processing of the propeptide, which is achieved in most cases intracellularly by the protease caspase-1 [3]. However, using caspase-1-deficient mice it has been shown that under certain conditions such as in neutrophil-dependent inflammation, other proteases than caspase-1 are also able to cleave and activate proIL-1β [14–15]. Nevertheless, caspase-1 is considered to be the master protease required for activation of IL1-β [3]. In contrast to proIL-1β, proIL-1α is able to bind and activate IL-1RI although proteolysis of its prosequence can enhance its biological activity [16]. Cell lysis upon injury or trauma therefore generates IL-1 activity due to passive release of proIL-1α, whereas passively released proIL-1β does not possess biological activity.
Most secreted proteins contain a signal peptide at the amino terminus, which directs them through the classical endoplasmic reticulum/Golgi-dependent secretion pathway to the extracellular space. In contrast, proIL-1α, proIL-1β and several other proteins are leaderless, thus lacking a signal peptide, and are secreted by a non-canonical pathway called the unconventional protein secretion pathway [17]. The mechanism and regulation of unconventional secretion is only poorly understood but it has recently been demonstrated that caspase-1 activity is required for the secretion of leaderless proteins [18–19].
As mentioned above, IL-1Ra blocks IL-1 activity. Although IL-1Ra, IL-1α and IL-1β have a similar affinity to IL-1RI, a large excess (100–1,000) of IL-1Ra over IL-1 is required for efficient blockade of receptor signalling in vitro and in vivo [20].
Inflammasomes and regulation of IL-1β activation
When proIL-1β synthesis is induced in circulating monocytes, mature IL-1β is secreted because these cells contain small amounts of active caspase-1 [21]. However, in all other cell types caspase-1 is inactive in the absence of activation signals, since this protease is synthesised as an inactive proenzyme. Activation of the protease takes place in recently identified innate immune complexes called inflammasomes [3]. The NLRP1 (nucleotide-binding domain, leucine-rich repeat-containing receptor protein, also called NALP1) inflammasome was identified in 2002 in a cell-free system of the myeloid cell line THP1 [22]. NLRP1 is the large backbone protein of the complex, which upon assembly of the inflammasome complex binds to caspase-5 and to ASC (apoptosis-associated speck-like protein containing a caspase recruitment domain (CARD)). The latter interacts with caspase-1, which brings both proteases into close proximity, resulting in their activation [22] (fig. 1). NLRP3, NLRP6 and AIM2 (absent in melanoma 2) can also bind caspase-1 via binding to the adaptor protein ASC, whereas NLRC4 (Nod-like receptor family CARD domain-containing proteins) interacts directly with caspase-1 [23]. Assembly of these complexes results in oligomerisation and subsequent activation of caspase-1. As a consequence of this, active caspase-1 cleaves and activates proIL-1β, but also the proinflammatory cytokine proIL-18.
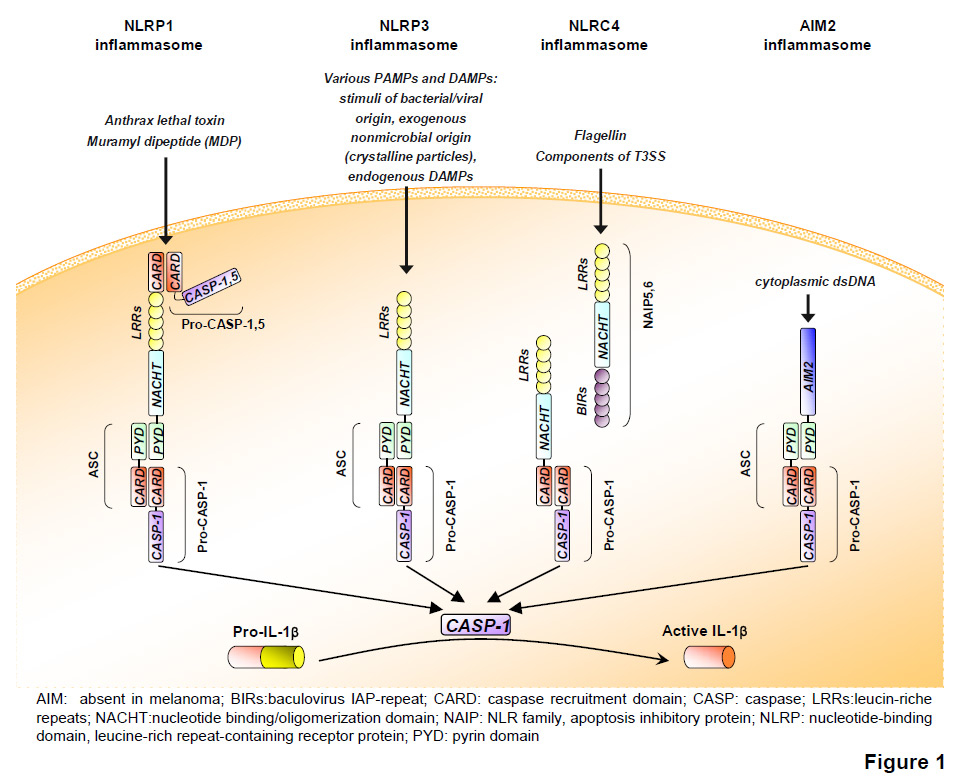
Figure 1
The inflammasomes. Several types of inflammasomes have been described. Upon exposure to specific PAMPs/DAMPs, respective inflammasomes are activated, leading to the proteolytic cleavage of caspase-1 or -5 (CASP) and subsequent cleavage of pro-IL1β into its active, secreted form.
Inflammasomes mediate the assembly and activation of caspase-1 in response to selected proinflammatory stimuli, including pathogen-associated molecular patterns (PAMPs) such as LPS and endogenous damage-associated molecular patterns (DAMPs) such as ATP. Four types of inflammasome complex that are activated upon encounter with distinct types of DAMPs and/or PAMPs have been characterised. These are the Aim2, NLRP1, NLRP3 and NLRC4 inflammasomes (fig. 1). The Aim2 inflammasome is activated by binding to viral and bacterial double-stranded DNA resulting from intracellular pathogens, the NLRP1 inflammasome by muramyl dipeptide, and NLRC4 inflammasome by flagellin [23]. The NLRP3 inflammasome is very probably the most important inflammasome since it assembles in response to a large variety of PAMPs and DAMPs, and its deficiency is already clearly associated with immunological dysfunction in mice as well as disease states in man. Since direct interaction of any of these activators with NLRP3 could not be shown, indirect activation mechanisms have been suggested. Particulates such as asbestos or crystals such as gout-causing monosodium urate (MSU) or cholesterol are phagocytosed by macrophages [24–25]. However, their clearance is not efficient and this results in lysosomal rupture, which, in turn, activates the NLRP3 inflammasome by an unknown mechanism possibly involving cathepsins [25–26]. As all NLRP3 activators induce the generation of reactive oxygen species (ROS), it has been reported that this ROS production triggers NLRP inflammasome activation [26]. Generation of ROS induces dissociation of thioredoxin from thioredoxin-interacting protein. Then the latter is able to bind NLRP3, which induces inflammasome activation [27]. The predominant source of ROS generated by danger signals is most probably the mitochondria, which also controls inflammasome activation via the release of mitochondrial DNA [28–29].
Inflammasome-dependent activation of caspase-1 induces not only secretion of IL-1β and of IL-18 but also a lytic form of cell death called pyroptosis [30–32]. In pyroptotic cells caspase-1-dependent DNA fragmentation occurs, though this does not require classical apoptotic caspases. Pyroptosis is characterised by osmotic swelling of the cell, which results in rupture of the plasma membrane. This induces a strong inflammatory response through the release of proinflammatory cytokines such as proIL-1α, but also via spreading of ATP or other proinflammatory molecules [33]. In contrast, apoptosis is immunologically silent.
The skin
At the body`s surface the skin prevents the loss of essential body fluids and invasion of microbes and irritants. Its outer layer is the epidermis, which consists mainly of keratinocytes and contains few Langerhans cells, a specialised form of DC (dentritic cells), and some pigment-producing melanocytes [4–5]. Keratinocytes express several types of keratin, proteins, which are expressed only in epithelia, form intermediate filaments through assembly into bundles and generate the toughness of the epidermis [34]. In the underlying dermis several types of immune cell can be found, such as macrophages, T cells and mast cells. They are embedded in connective tissue, which is made up by a mixture of extracellular matrix proteins produced by fibroblasts. The supply of nutrients of the whole skin is guaranteed by blood vessels in the dermis, the latter also containing lymphatic vessels and nerve endings. In contrast to the dermis, the epidermis is a constantly renewing tissue. Proliferation of keratinocytes is restricted to the basal cell layer, where stem cells and transit amplifying cells are located. During an apoptosis-like process of terminal differentiation, keratinocytes change their expression profile and properties and migrate to the outer surface of the epidermis. This generates several densely-packed layers of keratinocytes with dead, flat and keratin-filled corneocytes at the surface, which form a protective envelope.
Under homoeostatic conditions, the skin is colonised by a certain number and diversity of microorganisms on its surface. The dynamic equilibrium between the epidermis and microorganisms is regulated inter alia by sebocytes, which are located in sebaceous glands within the dermis and produce anti-microbial lipids, as well as by the microbes themselves, which produce antibiotic and antifungal substances as well as bacteriolytic enzymes. In addition, keratinocytes express antimicrobial substances constitutively and upon injury or infection, e.g. by stimulation of TLRs through PAMPs and DAMPs [35]. Certain keratinocyte-derived antimicrobial peptides as well as cytokines can influence the immunological properties of dendritic cells and T cells, which are also influenced by stimulation of their own TLRs and can activate adaptive immunity [4–5]. Therefore, on the one hand the skin must ensure an efficient defence against pathogens and immunosurveillance, and on the other hand the skin must minimise excessive immune responses which can result in disease states such as allergy, chronic inflammation and autoimmunity.
IL-1 and inflammasomes in keratinocytes
In contrast to macrophages and DCs, human primary keratinocytes constitutively express inflammasome proteins as well as proIL-1α, proIL-1β, and IL-1Ra [36–37]. Irradiation with a physiological dose of UVB induces secretion of proIL-1α and of mature and active IL-1β comparable to the amount released by activated macrophages [36]. This secretion requires expression of NLRP1, NLRP3, ASC, caspase-4 and caspase-1, as well as caspase-1 and -4 activity [38]. Assembly of the inflammasome is dependent on an increase in the concentration of cytoplasmic Ca2+. Most importantly, caspase-1 expression is also required in vivo in mice for the induction of an inflammatory response upon UVB irradiation [36]. This points to an important role of keratinocytes in the induction of sunburn and in innate immune responses dependent on the inflammasome and IL-1.

Figure 2
Current models of inflammasome activation involve K+ efflux, release of reactive oxygen species (ROS) and mitochondrial DNA (mtDNA) in the cytosol. Upon encounter of PAMPs/DAMPs including microbes or crystals, the NLRP3–inflammasome is activated and the resulting IL-1β release triggers localised inflammatory responses. In contrast, the NLRP3–inflammasome is constitutively activated, resulting in multi-organ involvement and periodic fever in patients suffering from autoinflammatory disorders.
Contact hypersensitivity (CH) is a frequent inflammatory skin disease, also called eczema, which can be studied in established mouse models of allergic contact dermatitis and is based on an adaptive immune response to haptens [37]. In the first phase, application of haptens to the skin primes T cells. A second hapten stimulus induces a local inflammatory response. This inflammation is most probably dependent on the migration of Langerhans cells to lymph nodes, subsequent antigen presentation and recruitment of expanded T cells to the site of hapten application [5, 39]. Recent evidence provided by our group and others has now shown that the earliest phase of sensitisation in CH critically depends on IL-1, ASC and NLRP3 expression and can be inhibited by administration of IL-1Ra [37, 40–42]. IL-1 and the inflammasome is involved in sensing contact sensitizers in the skin and triggering an innate immune response to these, which subsequently also very probably plays a role in helping trigger the adaptive immune response to contact sensitisers in the skin.
Keratinocytes express in vitro and in vivo the inflammasome component Aim2, a molecule which is important for sensing intracellular double stranded DNA. Accordingly, transfection of keratinocytes with double-stranded DNA in vitro induces secretion of mature IL-1β in an Aim2- and caspase-1-dependent manner [43–44]. Whether Aim2 expression is increased in skin biopsies of patients suffering from psoriasis, a chronic inflammatory skin disease, is controverted. Interestingly, cytosolic DNA was detected in keratinocytes of psoriatic lesions in contrast to healthy skin, suggesting that cytosolic DNA may act as a DAMP in psoriasis via activation of the Aim2 inflammasome [43]. The antimicrobial peptide LL-37, which can interact with DNA and induce interferon alpha production by plasmacytoid dendritic cells in psoriatic skin, can also block activation of the Aim2 inflammasome by binding to and neutralising cytosolic DNA. These data suggest that the generation of IL-1 activity by the Aim2 inflammasome in keratinocytes may contribute to the pathogenesis of psoriasis.
An important illustration of the consequences of dysregulated control of inflammasome activation for the skin and inflammatory disease was recently provided by experiments in mice where the NLRP3 gene mutation causing CAPS (cryopyrin-associated periodic syndromes) was introduced into mice. These mice that have a hyperactive inflammasome spontaneously develop a neutrophil rich inflammation of the skin with a TH17 dominant immune response [45]. Interestingly, a redundant feature of inflammasome activation and IL-1β release in tissues thus appears to be the neutrophilic nature of the inflammatory infiltrates at sites of tissue inflammation. This feature is also observed in several human autoinflammatory diseases.
Autoinflammatory diseases with skin involvement
Autoinflammatory diseases are a relatively new category of diseases distinct from allergic and autoimmune diseases and characterised by seemingly unprovoked recurrent inflammation in the absence of evidence of circulating autoantiobodies or an antigen-specific T cell response [46] (fig. 2). Recently it has been shown that several mutations in proteins of the inflammasome complex or proteins that regulate the function of the inflammasome are associated with autoinflammatory disease, including the cryopyrin-associated periodic syndromes such as Muckle-Wells syndrome, but also other diseases such as Schnitzler’s syndrome and Behçet’s disease in which IL-1 also appears to play a critical role.
|
Table 1: Autoinflammatory disorders with skin involvement. |
| Disease |
Affected tissues |
Deficient molecule |
IL-1–targeting treatment |
Target molecule |
|
CAPS
|
|
FCAS
|
Skin, joints, eyes |
NLRP3 |
Anakinra
Rilonacept
Canakinumab |
IL-1R1
IL-1β
IL-1β |
|
MWS
|
Skin, joints, eyes, ears, meninges |
NLRP3 |
Anakinra
Rilonacept
Canakinumab |
IL-1R1
IL-1β
IL-1β |
|
CINCA
|
Skin, joints, eyes, ears, meninges and bones |
NLRP3 |
Anakinra
Canakinumab |
IL-1R1
IL-1β |
|
Other inflammatory disorders
|
|
DIRA
|
Skin, bones, lungs,
blood vessels |
IL-1RA |
Anakinra |
IL-1R1 |
|
PAPA syndrome
|
Skin, joints |
PSTPIP1 |
Anakinra |
IL-1R1 |
|
Schnitzler syndrome
|
Skin, joints, bones |
? |
Anakinra |
IL-1R1 |
|
Sweet’s syndrome
|
Skin |
? |
Anakinra |
IL-1R1 |
|
Behcet’s disease
|
Skin, eyes, mucosa, gastro-intestinal tract, nervous system, blood vessels |
IL-1β |
Anakinra |
IL-1R1 |
Cryopyrin-associated periodic syndromes
Cryopirin-associated periodic syndromes (CAPS, OMIM/606416) are a group of rare inherited inflammatory disorders caused by different autosomal dominant mutations in the cryopyrin-coding gene NLRP3(nucleotide-binding domain, leucine-rich repeat containing gene family, pyrin domain containing protein 3) on chromosome 1q44, also called CIAS1, PYPAF1or NALP3. To date more than 90 mutations associated with CAPS have been reported. Such mutations lead to constitutive activation of caspase-1 and subsequent abnormal IL-1β secretion. CAPS consists of a spectrum of hereditary periodic fever syndromes that include familial cold autoinflammatory syndrome (FCAS; OMIM/120100), Muckle-Wells Syndrome (MWS; OMIM/191900), and chronic infantile neurological cutaneous and articular syndrome (CINCA; OMIM/607115), also known as neonatal-onset multisystem inflammatory disease (NOMID). Clinically periodic fever and urticaria-like skin lesions are hallmarks, and can be associated with one or more of the following depending on severity: arthritis, conjunctivitis, amyloidosis, sensorineural hearing loss, aseptic meningitis and/or cerebral atrophy.
Interleukin-1 blockade has shown very good therapeutic efficacy with rapid resolution of clinical symptoms and complete normalisation of inflammatory markers such as CRP. The recombinant IL-1 receptor antagonist anakinra is currently the most used medication for the treatment of CAPS. Canakinumab – a recombinant, fully human, monoclonal, anti-IL-1β antibody – and rilonacept – a dimeric fusion protein consisting of the extracellular domains of IL-1R1 and IL-1RAP joined to Fc domain of human IgG1 are effective alternative IL-1 inhibitors for this indication [47–48].
Familial cold autoinflammatory syndrome
FCAS is the mildest form of CAPS and typically starts in the first year of life. FCAS presents with recurrent attacks of a maculopapular, painful, non-pruritic urticarial exanthema accompanied by fever, chills, joint stiffness, conjunctivitis, and headache after exposure to cold [49]. The urticarial rash begins 1–3 hrs after cold stimulus, peaks at 2–6 hrs and is accompanied by sweating. This seemingly unprovoked sterile inflammation can result in neurosensory deafness, intellectual impairment and meningitis [50]. Amyloidosis develops in fewer than 5% of patients.
The urticarial exanthema in CAPS patients is similar to that observed in common urticaria. However, unlike in common urticaria, anti-IL-1 therapy – rather than antihistamines – results in complete remission of symptoms in patients suffering from FCAS as long as the treatment is given.
Muckle-Wells syndrome
Muckle-Wells syndrome (MWS), also known as urticaria-deafness-amyloidosis syndrome, has an intermediate severity among CAPS and was first reported in 1962. MWS is a hereditary disease related to FCAS with recurrent episodes of urticaria-like skin lesions appearing without cold exposure [51]. MWS is also associated with late-onset neurosensory deafness and renal amyloidosis occurring in 25% of patients.
Chronic infantile neurological cutaneous and articular syndrome
CINCA was first described in 1981 and is the most severe form of CAPS. It occurs sporadically without a family history and typically starts during the neonatal period. Symptoms of CINCA include high fever, maculopapular or urticaria-like, non-pruritic migratory exanthemas, arthropathy, lymphadenopathy, hepatosplenomegaly and chronic meningitis. CINCA is also associated with progressive loss of vision and neurosensory deafness, and mental and growth retardation. 20% of CINCA patients die within the first two decades.
De novo mutations in NLRP3 can be found in only 50–60% of patients [52–53]. Long-term efficacy of Kineret treatment of patients suffering from CINCA has been reported [54].
Other autoinflammatory diseases
Deficiency of interleukin-1 receptor antagonist
The recently reported deficiency of interleukin-1 receptor antagonist (DIRA; OMIM/147679) is an autosomal recessive inherited disease caused by mutations in IL1RN on chromosome 2 leading to absence of IL-1RA and subsequent IL-1 overactivity [55–56]. DIRA manifests itself clinically with perinatal-onset pustular dermatitis, joint swelling, painful osteolytic lesions and periosteitis. In contrast to CINCA, fever is rare and neurologic inflammation is absent.
Treatment with anakinra – substituting the missing protein in patients with its recombinant form – results in rapid clinical improvement [57].
Pyogenic arthritis-pyoderma gangrenosum-acne syndrome
Pyogenic arthritis-pyoderma gangrenosum-acne syndrome (OMIM/604416), known asPAPA syndrome, is a rare inherited disorder caused by an autosomal dominant mutation in PSTPIP1(proline-serine-threonine-phosphatase interactive protein 1) also known as CD2BP1(CD2-binding protein 1) on chromosome 15q24– q25.1 [58]. Although presenting different clinical features, the pathophysiologies of PAPA and FMF are related.
Sterile erosive arthritis especially of the knees, elbows and ankles, usually starts in early childhood. Severe cystic acne which can persist well into adulthood develops with puberty, while arthritic symptoms tend to regress. Pyoderma gangrenosum is variably expressed and manifests as poorly healing ulcers preferentially developing on the distal limbs but also in a multifocal fashion on the entire skin. In PAPA syndrome patients, the regulation of caspase-1 activation is lacking and increased production of IL-1β as well as tumour necrosis factor (TNF) in peripheral blood mononuclear cells has been reported [59–60].
The treatment of PAPA syndrome largely depends on the dominant clinical manifestation. Anakinra has been shown to be effective in controlling inflammatory lesions in PAPA syndrome patients [61–62]. Infliximab (anti-TNF) has been reported to be successful [63]. Arthritis episodes are usually successfully treated with cortisteroids. Pyoderma gangrenosum often responds poorly to systemic corticosteroids and is therefore usually treated with immunosuppressants.
Schnitzler syndrome
Schnitzler syndrome usually presents in mid-adulthood with non-pruritic urticaria-like exanthemas and IgM gammopathy variably accompanied by fever, arthritis or arthralgia, bone pain, lymphadenopathy and hepato- and/or splenomegaly [64]. Both the exact aetiology and pathogenesis of Schnitzler syndrome remain to be clarified.
Therapies using antihistamines, non-steroidal antiinflammatory drugs, corticosteroids, immunosuppressive drugs, colchicine, dapsone or thalidomide have shown very limited effects. TNF blockers have been shown to be ineffective. A mutation in NLRP3 has been reported in a single case, suggesting possible involvement of the inflammasome in the pathogenesis [65]. Enhanced IL-1 secretion by peripheral blood mononuclear cells has been demonstrated in Schnitzler’s syndrome, and although the cause of this has still to be identified, the recent evidence in favour of clinical efficacy of anakinra and canakinumab in this syndrome suggest that IL-1β plays an important role in this disease [66].
Sweet’s syndrome
Sweet’s syndrome was first reported in 1964 as an acute febrile neutrophilic dermatosis [67]. Sweet's syndrome (OMIM/608068) is a neutrophilic skin disease characterised by fever, an elevated neutrophil count, and painful erythematous cutaneous nodules or plaques constituted histologically by a diffuse dense infiltrate of mature neutrophils in the upper dermis. Sweet's syndrome is frequently associated with haematological malignancies or chronic inflammatory disorders such as rheumatoid arthritis and inflammatory bowel disease, but may also occur in the absence of coexisting disease and is to date considered a hypersensitivity reaction. A possible link with HLA-Bw54 is suspected in patients with Sweet’s syndrome, and high levels of the cytokine G-CSF (granulocyte-colony stimulating factor) have been demonstrated in the serum of patients. Furthermore, cases are reported where exogenous G-CSF has triggered the disease, suggesting that this cytokine, which promotes the production and survival of neutrophils, plays a role in the pathogenesis of Sweet’s syndrome. Sweet syndrome does fulfil the current criteria for classification as an autoinflammatory disease, notably the seemingly unprovoked inflammation in the absence of detectable autoantibodies and evidence of an antigen-specific T-cell response.
Systemic corticosteroid therapy remains the gold standard for the clinical management of patients with Sweet's syndrome. However, recently very encouraging responses to Kineret administration have been reported, suggesting that IL-1 and the inflammasome may play a significant role in the pathophysiology of this autoinflammatory disease [68–69].
Behçet's disease
Behçet's disease (BD, OMIM/109650) is a chronic systemic inflammatory disorder characterised by the following triad of symptoms: recurrent oral aphthous ulcers, genital ulcers, and uveitis [70]. BD is rare in Western countries but more frequent in the Middle and Far East. The clinical constellation of BD includes inflammatory lesions of mucocutaneous tissues (oral and genital ulceration, pseudofolliculitis, acne- and erythema nodosum-like skin lesions), as well as ocular, vascular, digestive, and/or nervous system tissues. The pathogenesis of BD is incompletely understood and is suggested to be of autoimmune aetiology, although strong evidence for a causative autoantigen and respective autoantibodies is lacking to date. Triggering of an immune reaction by certain infectious agents that may inhabit the oral cavity (Streptococcus and Staphylococcus species, herpes simplex virus (HSV) and/or Escherichia coli) has been suggested as playing a role in BD [71–72]. The systemic involvement of multiple organs, which can be observed in BD, is based histopathologically on the development of vasculitic lesions due to tissue infiltration with both T cells and neutrophils. The presence of neutrophils and the fact that BD is characterised by recurrent episodes of inflammation without apparent cause open up the possibility that BD may be an autoinflammatory disease. Interestingly, among the predisposing genetic factors that are considered to play an important role in the development of BD are specific IL-1β gene polymorphisms that have been reported to increase susceptibility to BD in the Turkish population [73–74]. Furthermore, as in several diseases considered to be autoinflammatory to date, excellent clinical responses have been reported in BD patients treated with Kineret, reinforcing the possibility of a role for IL-1 in the pathogenesis of BD [75–76].
Conclusion
While being a key player in innate immune responses, recent evidence has clearly demonstrated that in certain instances IL-1β overproduction may be the cause, or be associated with several more or less severe inflammatory diseases, several of which have recently been classified as autoinflammatory diseases and are associated with skin lesions. This relatively new group of diseases is considered to be distinct from autoimmune and allergic diseases, in that both the presence of autoantibodies and evidence of an antigen-specific T-cell response which characterise the latter are absent in the former. The discovery of the inflammasomes as key intracellular sensing complexes required for IL-1 secretion has opened the way to elucidation of the pathomechanisms involved in autoinflammation, certain autoinflammatory diseases, and several inflammatory skin diseases caused by external environmental factors. These diseases have in common their recurrent inflammatory nature, multisytemic involvement, the skin as a frequently involved organ, and the frequent contribution of neutrophils to the inflammatory lesions observed. The new understanding of the role of the inflammasome and IL-1 in an emerging set of diseases has led the successful development and implementation in clinical trials of therapies targeted against IL-1 for autoinflammatory diseases, prime examples of which are the use of IL-1 antagonists in cryopyrin-associated periodic syndromes such as Muckle-Wells syndrome.
References
1 Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454(7203):428–35.
2 May SA, et al. Inflammation: a clinical perspective. The Ciba-Geigy Prize for Research in Animal Health. Vet Rec. 1987;120(22):514–7.
3 Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol. 2009;27:229–65.
4 Feldmeyer L, et al. Interleukin-1, inflammasomes and the skin. Eur J Cell Biol. 2010;89(9):638–44.
5 Nestle FO, et al. Skin immune sentinels in health and disease. Nat Rev Immunol. 2009;9(10):679–91.
6 Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–50.
7 Dinarello CA. Blocking interleukin-1beta in acute and chronic autoinflammatory diseases. J Intern Med. 2011;269(1):16–28.
8 Weber A, Wasiliew P, Kracht M. Interleukin-1 (IL-1) pathway. Sci Signal. 2010;3(105): p. cm1.
9 Dinarello CA. Interleukin-1, interleukin-1 receptors and interleukin-1 receptor antagonist. Int Rev Immunol. 1998;16(5–6):457–99.
10 Dinarello CA. Therapeutic strategies to reduce IL-1 activity in treating local and systemic inflammation. Curr Opin Pharmacol. 2004;4(4):378–85.
11 Hacham M, et al. Different patterns of interleukin-1alpha and interleukin-1beta expression in organs of normal young and old mice. Eur Cytokine Netw. 2002;13(1):55–65.
12 Schindler R, Clark BD, Dinarello CA. Dissociation between interleukin-1 beta mRNA and protein synthesis in human peripheral blood mononuclear cells. J Biol Chem. 1990;265(18):10232–7.
13 Schindler R, Gelfand JA, Dinarello CA. Recombinant C5a stimulates transcription rather than translation of interleukin-1 (IL-1) and tumor necrosis factor: translational signal provided by lipopolysaccharide or IL-1 itself. Blood. 1990;76(8):1631–8.
14 Guma M, et al. Caspase 1–independent activation of interleukin-1beta in neutrophil-predominant inflammation. Arthritis Rheum. 2009;60(12):3642–50.
15 Joosten LA, et al. Inflammatory arthritis in caspase 1 gene-deficient mice: contribution of proteinase 3 to caspase 1–independent production of bioactive interleukin-1beta. Arthritis Rheum. 2009;60(12):3651–62.
16 Afonina IS, et al. Granzyme B-dependent proteolysis acts as a switch to enhance the proinflammatory activity of IL-1alpha. Mol Cell. 2011;44(2):265–78.
17 Nickel W. The mystery of nonclassical protein secretion. A current view on cargo proteins and potential export routes. Eur J Biochem. 2003;270(10):2109–19.
18 Keller M, et al. Active caspase-1 is a regulator of unconventional protein secretion. Cell. 2008;132(5):818–31.
19 Nickel W, Rabouille C. Mechanisms of regulated unconventional protein secretion. Nat Rev Mol Cell Biol. 2009;10(2):148–55.
20 Gabay C, Lamacchia C, Palmer G. IL-1 pathways in inflammation and human diseases. Nat Rev Rheumatol. 2010;6(4):232–41.
21 Netea MG, et al. Differential requirement for the activation of the inflammasome for processing and release of IL-1 beta in monocytes and macrophages. Blood. 2009;113(10):2324–35.
22 Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10(2):417–26.
23 Strowig T, et al. Inflammasomes in health and disease. Nature. 2012;481(7381):278–86.
24 Duewell P, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464(7293):1357–U7.
25 Hornung V, Latz E. Critical functions of priming and lysosomal damage for NLRP3 activation. Eur J Immunol. 2010;40(3):620–3.
26 Tschopp J, Schroder K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat Rev Immunol. 2010;10(3):210–5.
27 Zhou R, et al. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11(2):136–40.
28 Nakahira K, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12(3):222–30.
29 Shimada K, et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity, 2012.
30 Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7(2):99–109.
31 Ting JP, Willingham SB, Bergstralh DT. NLRs at the intersection of cell death and immunity. Nat Rev Immunol. 2008;8(5):372–9.
32 Yeretssian G, Labbe K, Saleh M. Molecular regulation of inflammation and cell death. Cytokine. 2008;43(3):380–90.
33 Miao EA, et al. Caspase-1–induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol. 2010;11(12):1136–42.
34 Fuchs E, Raghavan S. Getting under the skin of epidermal morphogenesis. Nat Rev Genet. 2002;3(3):199–209.
35 Glaser R, et al. Antimicrobial psoriasin (S100A7) protects human skin from Escherichia coli infection. Nat Immunol. 2005;6(1):57–64.
36 Feldmeyer L, et al. The inflammasome mediates UVB-induced activation and secretion of interleukin-1beta by keratinocytes. Curr Biol. 2007;17(13):1140–5.
37 Watanabe H, et al. Activation of the IL-1beta-processing inflammasome is involved in contact hypersensitivity. J Invest Dermatol. 2007;127(8):1956–63.
38 Sollberger G, et al. Caspase-4 is required for activation of inflammasomes. J Immunol. 2012;188(4):1992–2000.
39 O’Leary JG, et al. T cell- and B cell-independent adaptive immunity mediated by natural killer cells. Nat Immunol. 2006;7(5):507–16.
40 Kondo S, et al. Interleukin-1 receptor antagonist suppresses contact hypersensitivity. J Invest Dermatol. 1995;105(3):334–8.
41 Shornick LP, et al. Mice deficient in IL-1beta manifest impaired contact hypersensitivity to trinitrochlorobenzone. J Exp Med. 1996;183(4):1427–36.
42 Sutterwala FS, et al. Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity. 2006;24(3):317–27.
43 Dombrowski Y, et al. Cytosolic DNA triggers inflammasome activation in keratinocytes in psoriatic lesions. Sci Transl Med. 2011;3(82):82ra38.
44 Kopfnagel V, Wittmann M, Werfel T. Human keratinocytes express AIM2 and respond to dsDNA with IL-1beta secretion. Exp Dermatol. 2011;20(12):1027–9.
45 Meng G, et al. A mutation in the Nlrp3 gene causing inflammasome hyperactivation potentiates Th17 cell-dominant immune responses. Immunity. 2009;30(6):860–74.
46 Masters SL, et al. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease (*). Annu Rev Immunol. 2009;27:621–68.
47 Hoffman HM, et al. Efficacy and safety of rilonacept (interleukin-1 Trap) in patients with cryopyrin-associated periodic syndromes: results from two sequential placebo-controlled studies. Arthritis Rheum. 2008;58(8):2443–52.
48 Kuemmerle-Deschner JB, et al. Two-year results from an open-label, multicentre, phase III study evaluating the safety and efficacy of canakinumab in patients with cryopyrin-associated periodic syndrome across different severity phenotypes. Ann Rheum Dis. 2011;70(12):2095–102.
49 Glaser RL, Goldbach-Mansky R. The spectrum of monogenic autoinflammatory syndromes: understanding disease mechanisms and use of targeted therapies. Curr Allergy Asthma Rep. 2008;8(4):288–98.
50 Goldbach-Mansky R, et al. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. N Engl J Med. 2006;355(6):581–92.
51 Hawkins PN, et al. Spectrum of clinical features in Muckle-Wells syndrome and response to anakinra. Arthritis Rheum. 2004;50(2):607–12.
52 Agostini L, et al. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity. 2004;20(3):319–25.
53 Aksentijevich I, et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum. 2002;46(12):3340–8.
54 Neven B, et al. Long-term efficacy of the interleukin-1 receptor antagonist anakinra in ten patients with neonatal-onset multisystem inflammatory disease/chronic infantile neurologic, cutaneous, articular syndrome. Arthritis Rheum. 2010;62(1):258–67.
55 Aksentijevich I, et al. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N Engl J Med. 2009;360(23):2426–37.
56 Reddy S, et al. An autoinflammatory disease due to homozygous deletion of the IL1RN locus. N Engl J Med. 2009;360(23):2438–44.
57 Schnellbacher C, et al. Deficiency of Interleukin-1 Receptor Antagonist Responsive to Anakinra. Pediatr Dermatol. 2012.
58 Nesterovitch AB, et al. Mutations in the PSTPIP1 gene and aberrant splicing variants in patients with pyoderma gangrenosum. Clin Exp Dermatol. 2011;36(8):889–95.
59 Cortis E, et al. Abnormal production of tumor necrosis factor (TNF) – alpha and clinical efficacy of the TNF inhibitor etanercept in a patient with PAPA syndrome [corrected]. J Pediatr. 2004;145(6):851–5.
60 Shoham NG, et al. Pyrin binds the PSTPIP1/CD2BP1 protein, defining familial Mediterranean fever and PAPA syndrome as disorders in the same pathway. Proc Natl Acad Sci U S A. 2003;100(23):13501–6.
61 Brenner M, et al. Targeted treatment of pyoderma gangrenosum in PAPA (pyogenic arthritis, pyoderma gangrenosum and acne) syndrome with the recombinant human interleukin-1 receptor antagonist anakinra. Br J Dermatol. 2009;161(5):1199–201.
62 Dierselhuis MP, et al. Anakinra for flares of pyogenic arthritis in PAPA syndrome. Rheumatology (Oxford). 2005;44(3):406–8.
63 Stichweh DS, Punaro M, Pascual V. Dramatic improvement of pyoderma gangrenosum with infliximab in a patient with PAPA syndrome. Pediatr Dermatol. 2005;22(3):262–5.
64 Asahina A, et al. Schnitzler’s syndrome with prominent neutrophil infiltration misdiagnosed as Sweet’s syndrome: a typical example of urticarial neutrophilic dermatosis. Clin Exp Dermatol. 2010;35(4):e123–6.
65 Loock J, et al. Genetic predisposition (NLRP3 V198M mutation) for IL-1-mediated inflammation in a patient with Schnitzler syndrome. J Allergy Clin Immunol. 2010;125(2):500–2.
66 de Koning HD, et al. Successful canakinumab treatment identifies IL-1beta as a pivotal mediator in Schnitzler syndrome. J Allergy Clin Immunol. 2011;128(6):1352–4.
67 Sweet RD. An Acute Febrile Neutrophilic Dermatosis. Br J Dermatol. 1964;76:349–56.
68 Delluc A, et al. Efficacy of anakinra, an IL1 receptor antagonist, in refractory Sweet syndrome. Ann Rheum Dis. 2008;67(2):278–9.
69 Kluger N, et al. Efficacy of anti-interleukin-1 receptor antagonist anakinra (Kineret(R)) in a case of refractory Sweet’s syndrome. Dermatology. 2011;222(2):123–7.
70 Mendes D, et al. Behcet’s disease – a contemporary review. J Autoimmun. 2009;32(3–4):178–88.
71 Direskeneli H. Behcet’s disease: infectious aetiology, new autoantigens, and HLA-B51. Ann Rheum Dis. 2001;60(11): p. 996–1002.
72 Lehner T. The role of heat shock protein, microbial and autoimmune agents in the aetiology of Behcet’s disease. Int Rev Immunol. 1997;14(1):21–32.
73 Coskun M, et al. Specific interleukin-1 gene polymorphisms in Turkish patients with Behcet’s disease. Exp Dermatol. 2005;14(2):124–9.
74 Ozcimen AA, et al. IL-1 cluster gene polymorphisms in Turkish patients with Behcet’s disease. Int J Immunogenet. 2011;38(4):295–301.
75 Bilginer Y, Ayaz NA, Ozen S. Anti-IL-1 treatment for secondary amyloidosis in an adolescent with FMF and Behcet’s disease. Clin Rheumatol. 2010;29(2):209–10.
76 Botsios C, et al. Resistant Behcet disease responsive to anakinra. Ann Intern Med. 2008;149(4):284–6.