Aryl hydrocarbon receptor controls regulatory CD4+T cell function
DOI: https://doi.org/10.4414/smw.2012.13592
Summary
The ligand activated transcription factor aryl hydrocarbon receptor (AhR) has been studied for many decades in toxicology as the ligand for the environmental contaminant dioxin. However, AhR has recently emerged as a critical physiological regulator of immune responses affecting both innate and adaptive systems, and several AhR ligands with different pharmacological profiles have recently been studied. The current review discusses new insights into the role of AhR signalling and AhR ligands on the regulation of the immune system, with a focus on regulatory T cells which maintain immune tolerance. Notably, AhR is expressed and modulates the development of two induced regulatory CD4+ T cell subsets, the forkhead box P3-positive (Foxp3+) regulatory T cells (iTreg) and the IL-10-secreting type 1 regulatory T (TR1) cells, through different signalling pathways. We will finally discuss how AhR ligands could be exploited to alleviate human autoimmune diseases. Clearly, drugs targeted against AhR should promote the development of new strategies to fight against autoimmune diseases.
Introduction
The aryl hydrocarbon receptor (AhR) has been studied for its role in mediating toxicity of environmental contaminants such as dioxin. It is increasingly recognised that environmental toxins modulate immune responses and can both impair response to infection or trigger autoimmunity. AhR has emerged over the last decade as a critical regulator of the immune system. AhR is expressed both in cells involved in the innate immune response such as the dendritic cells (DCs) [1] or the innate lymphoid cells (ILC) [2], and in CD4+T cells which are crucial for adaptive immunity. Besides TH1 and TH2 subsets described by Mosmann and Coffman in 1986 [3], CD4+ T cells can differentiate in the periphery into additional subsets including pro-inflammatory effector T cells (TH17, TH22) and regulatory T cells (forkhead box P3 transcription factor (Foxp3)+ iTregs, type 1 regulatory T (TR1) cells or TH3) (reviewed in [4, 5]). AhR is expressed at high levels in two subsets of regulatory T cells (Tregs), the iFoxp3+Tregs and TR1 cells, as wells in TH17 [6–10]. This expression profile suggests that AhR plays an important role in regulatory T cells.
The immune system has evolved to protect organisms from infection but needs to avoid harmful responses to itself. In this regard, regulatory CD4+ T cells are crucial to maintain tolerance. Although suppressor T cells were described in the early 1970s, interest in regulatory T cells has been revived thirty years later with the discovery of the “lineage specific” transcription factor Foxp3 [11, 12]. Foxp3+CD4+Treg cells have emerged as critical regulators of the immune system as illustrated by the severe autoimmune inflammation observed in mice deficient in Foxp3 [13] or in patients with dysfunctional FOXP3 protein [14]. Foxp3+Tregs can be divided into thymus-derived natural Tregs (nTregs) that express the α chain of the IL-2 receptor CD25 and into inducible CD4+Tregs cells which are generated from CD25– precursors in the peripheral lymphoid organs (iTregs) (reviewed in [15]). Aside from iTregs, additional inducible regulatory CD4+T cells have been described including TGF-β-secreting TH3 [16] and type-1 regulatory T-cells (TR1) [17]. Despite their common role in the regulation of immune responses, induced regulatory T-cell subsets feature differences in their biology, such as the cytokines that induce them or the mechanisms by which they mediate suppression (table 1). Both iTregs and TR1 cells express AhR at a high level. The role of AhR signalling and the molecular basis downstream AhR activation differ between these two cell types, which will be discussed in more detail.
|
Table 1: Characteristics regulatory T cells.
CD4+ regulatory T cells can be divided in natural Foxp3+ Tregs which are generated in the thymus (nTregs) or Tregs generated in the periphery that comprise iFoxp3+Tregs, TR1 and TH3 cells.
CTLA4, Cytotoxic T-Lymphocyte Antigen 4; GITR, glucocorticoid-induced TNF receptor family-regulated gene; Tbx21, T-box transcription factor TBX21; LAP, latency-associated peptide. |
| |
nTregs
|
iTregs
|
TR1
|
TH3
|
| Place of generation |
Thymus |
Peripheral lymphoid organs |
Peripheral lymphoid organs |
Peripheral lymphoid organs |
| Differentiating cytokine |
– |
TGF-β |
IL-27 |
Anti-CD3 (oral tolerance) |
| Surface markers |
CD25 (IL-2R), CTLA4, GITR |
CD25 (IL-2R), CTLA4, GITR |
Unknown |
LAP
CD25- |
| Transcription factors |
Foxp3 |
Foxp3, AhR |
AhR, c-Maf, Tbx21 |
? |
| Growth promoting cytokine |
IL-2 |
IL-2 |
IL-21 |
? |
| Mode of suppression |
Multiple |
Multiple |
Multiple |
Multiple |
| Contact dependant |
CD39 |
CD39 |
Granzyme B |
LAP |
| Contact independent |
TGF-β, IL-35, IL-10 |
TGF-β, IL-35, IL-10 |
IL-10 |
TGF-β, IL-10 |
AhR ligands modulate immune response
AhR is a transcription factor that necessitates activation by a ligand to mediate its transcriptional activity. AhR reside in an inactive form in the cytosol in a complex composed of several proteins (Hsp90 or ARA9). Upon activation with a ligand, AhR undergoes conformational changes that enable its translocation to the nucleus. AhR can then initiate the transcription of promoters containing a dioxin-responsive element (DRE) consensus sequence. DRE are found in multiple gene promoters involved in the immune system.
AhR ligands can be divided into compounds of (1.) exogenous origin from environmental contaminants (man-made aromatic environmental pollutants or pharmaceutical components) or from natural origin (ligands synthetised by microbes or plants that can be found for example in food) and of (2.) endogenous ligands present in the human body (table 2). The ligand 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) is the most studied exogenous ligand and the greatest understanding of AhR biology are based on its effect. In vivo treatment with TCDD attenuates the disease course of several murine models of autoimmune diseases including multiple sclerosis, [7, 10] colitis [18] and uveoretinitis [19]. However, because of the unfavourable biological profile of TCDD (high affinity for AhR, long half-life) and implications in embryogenesis and tumourigenesis, its implementation in clinics is limited [20].
Besides environmental contaminants, natural AhR ligands are present in the diet or can be generated in the gastrointestinal tract from dietary compounds such as indirubin, a component found in traditional Chinese herbal medicine [21, 22], curcumin, a common spice used in Indian cuisine, or indole-3-carbinol found in cruciferous vegetables (table 2). Interests in natural AhR ligands and gut immunology are rising as illustrated by several recent publications. A first study pointed out that exposure to AhR ligands through the diet even in the first weeks of life is critical for the development of immune responses as they control the maturation of innate lymphoid cells (ILC) [2]. ILC drive immune responses against intestinal infections and their generation is impaired in AhR-deficient mice. Mice fed with diets lacking natural AhR ligands suffer from deficient ILC generation and are prone to intestinal infection. The sole addition of the natural AhR ligand indole-3-carbinol (table 2) in the diet restores both the generation of ILC and the immune response in an AhR-dependent manner. This highlights the importance of exposure to AhR agonists through food intake and their role in the maintenance of intestinal homeostasis. To corroborate those results, the group of Zhou similarly showed that AhR is necessary for the development of a subset of ILC (that are RORγt+) in mice [23]. Adult AhR-deficient mice have reduced numbers of ILC in both the small and large intestine, and are more susceptible to gut infections. Strikingly the deficits in ILC became evident when AhR-deficient mice were close to weaning age pointing towards the implication of exogenous factors such as food intake in the development of the immune response. The authors proposed that IL-22 production induced by AhR signalling is an important mediator for AhR protective effects in the gut, as IL-22 expression could rescue AhR-deficient mice from succumbing to severe gut infection. The group of Colonna obtained similar results and showed that AhR drives the development of ICL22 and postnatal lymphoid tissues which are critical for responding to gut infections [24]. They propose that AhR signalling regulates IL-22 partially through Notch pathways. Similarly, the group of Veldhoen showed that intra-epithelial lymphocytes (IELs), which are important in the first line defence against intestinal infection or skin defence, depend on AhR activation by dietary-derived ligands to maintain their generation and to control microbial load and composition in the gut [25]. Taken together, the results of those studies suggest that AhR signalling is important for the maintenance of gut immunity and also that exogeneous dietary AhR ligands are crucial to shape our immune response.
The recognition of endogenous ligands confers AhR a physiological role in fine-tuning the immune response and does not restrain AhR to be solely a receptor for environmental compounds. The ligand 6-formylindolo[3,2-b]carbazole (FICZ) was the first described endogenous AhR ligand (table 2). FICZ is formed when its precursor tryptophan is exposed to light in the skin. Similarly to TCDD, FICZ has immunoregulatory properties and administration of FICZ decreases the severity of colitis in different mouse models of inflammatory bowel disease (IBD) by reducing inflammatory responses and promoting the production of IL-22, an IL-10 family cytokine [18]. Another putative endogenous AhR ligand, the 2-(1'H-indole-3'-carbonyl)-thiazole-4-carboxylic acid methyl ester (ITE), induces functional Tregs that suppress EAE similarly to TCDD [26]. Recently, focus has been driven on the ligand kynurenine (Kyn), the first breakdown product in the indoleamine-2,3-dioxygenase (IDO)-dependent tryptophan, which is now increasingly recognised as an endogenous AhR ligand [9]. Indeed, Kyn is generated by dendritic cells (DCs) at the site of inflammation and promotes the generation of regulatory T cells akin to TCDD.
|
Table 2: AhR ligands.
Structures and origins of exogeneous, environmental pollutants and dietary, and endogeneous AhR ligands discussed in this review. |
|
AhR Ligands
|
Structure
|
Origin
|
|
Exogeneous ligands
|
|
Environmental pollutants
|
| – 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)
|
|
Toxic formed during combustion of organic compound |
|
Dietary
|
| – Indirubin |
|
Component of Chinese medicine |
| – Indol-3-carbinol |
|
Cruciferous vegetables
(broccoli) |
| – Curcumin
|
|
Indian spice |
|
Endogenous
|
| – 6-formylindolo[3,2-b]carbazole (FICZ) |
|
Tryptophan photoproduct |
| – 1'H-indole-3'-carbonyl)-thiazole-4 carboxylic acid methyl ester (ITE) |
|
Compound derived from indirubin |
| – Kynurenine (Kyn) |
|
Tryptophan product |
AhR and regulatory T cells
Funatake et al. first described the importance of AhR signalling in the generation of Tregs in vivo[27]. Using a mouse model of acute graft-versus-host response, they showed that donor T cells led to the generation of a subset of regulatory CD4+ T cells when transferred into mice treatedin vivo with TCDD. The induction of those regulatory T cells was AhR dependant and was abolished if recipient mice were deficient for AhR. The generation of those TCDD-induced Tregs did not results in an expansion of already circulating nTregs as TCDD-induced Tregs could be generated in a system devoid of nTregs. The authors pursued the characterisation of those TCDD-induced Tregs and performed an ex vivo characterisation of TCDD-induced CD4+ cells by comparing TCDD-induced Tregs with n-Tregs. They showed that TCDD-induced regulatory T cells were different from nTregs, expressed the surface markers CD25, CTLA4 and GITR and produced high levels of IL-10 and Granzyme B [6]. These pioneer in vivo studies provided the impetus to further study differentiation of T cells in vitro with purified T cells.
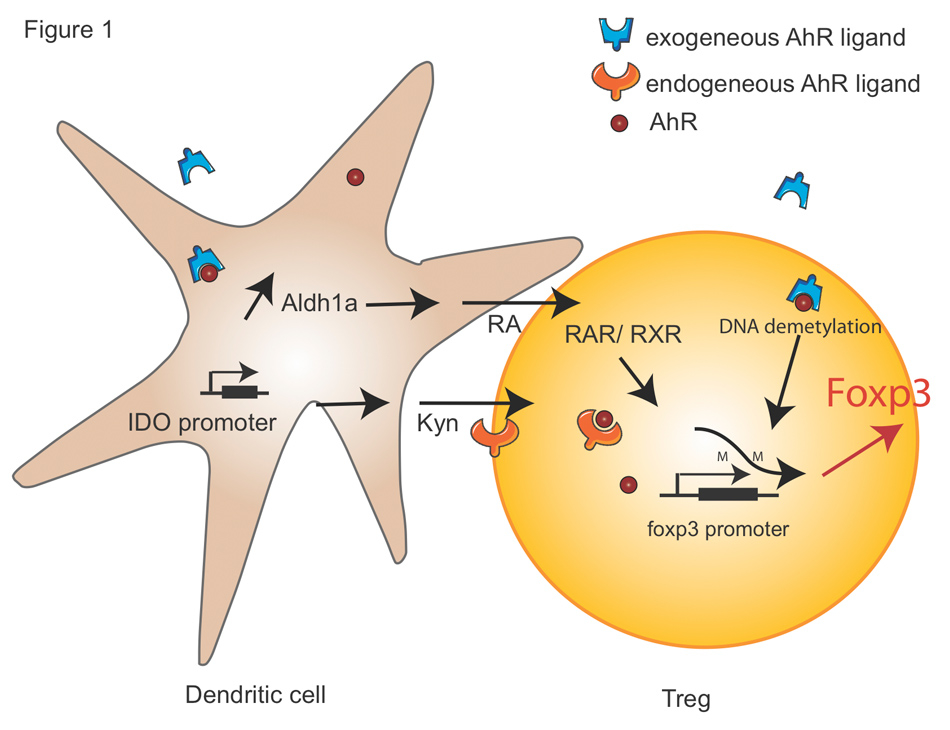
Figure 1
AhR signalling in Tregs.
The molecular mechanisms by which AhR promotes Foxp3+Tregs cell are shown. Exogenous AhR ligand induces tolerogenic DCs by multiple pathways. Firstly, the activation of AhR enhances the expression of Aldh1a1 which drives RA secretion. In T cells, RA binds to RAR and RXRs, which in turn bind to the Foxp3 promoter. Secondly, the activation of AhR induces the formation of Kyn in an IDO-dependant manner in DCs. Kyn further acts as an endogenously AhR ligand and promotes Foxp3 expression. In addition, the activation of AhR mediates partial demethylation of Foxp3 promoter in CD4+ T cells.
RA, retinoic acid; RAR, retinoic acid receptor; RXR, Retinoid X Receptor; Kyn, Kynurenine; IDO, Indoleamine 2,3-dioxygenase.
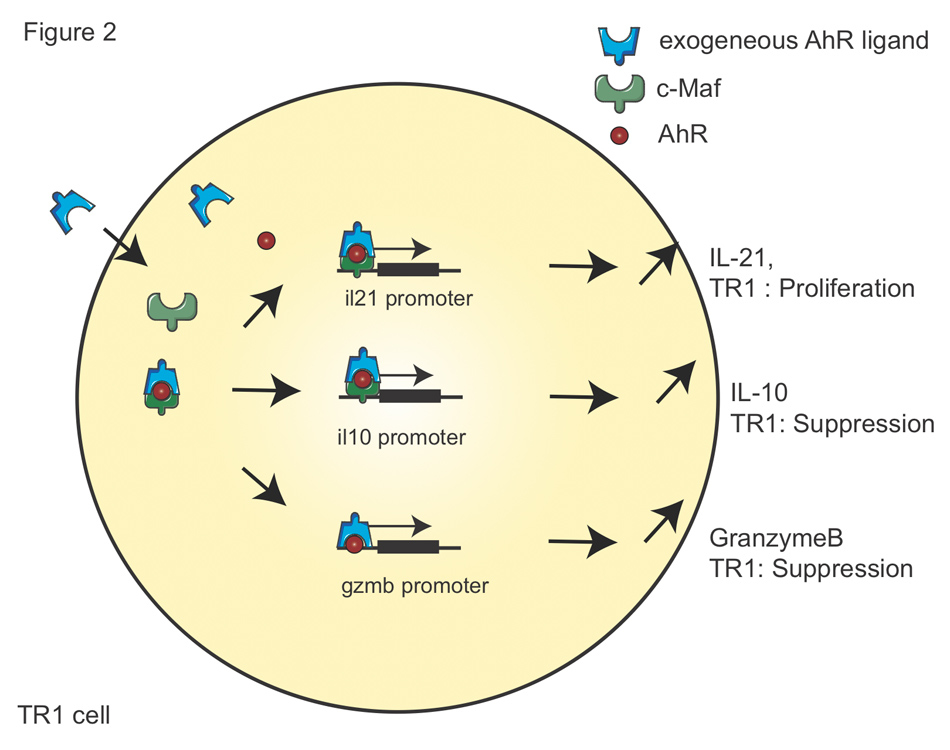
Figure 2
AhR signalling in TR1 cells.
The molecular mechanisms by which AhR promotes TR1 cell differentiation are shown. Firstly, AhR ligands bind to AhR and transactivates il21promoter with c-Maf that promotes TR1 cell proliferation. Secondly, the same complex of AhR and c-Maf transactivates il10promoter and mediates TR1 cell suppressive function. Thirdly, AhR drives the transactivation of Gzmb promoter that promotes granzyme B expression, which contributes to the suppressive activity of TR1 cells.
AhR and Foxp3+Tregs
As mentioned above, Foxp3+Tregs can be divided in thymus-derived nTregs and adaptive iTregs generated in the periphery. It has been proposed that in vivo TCDD do not enlarge nTregs population but promote the generation of iTregs in the periphery [27]. As early immuno-toxicological studies showed that exposure to TCDD contributes to thymic involution [28], one might think that exposure to TCDD impairs nTregs generation. The effect of TCDD or other AhR ligands on nTregs development in the thymus has not been formally studied as the analyses of TCDD-induced Tregs have been performed in adult mice.
TGF-β is crucial for the development of iTregs both in vitro and in vivo [29]. iTregs express high levels of CD25, the receptor of IL-2, and are dependent on an external IL-2 source to promote their growth (table 1). To study the role of AhR activation on Treg biology, numerous studies have been performed with the high affinity AhR agonist TCDD. Interestingly it was described many years ago that TCDD dampens effector T-cell function and promotes the development of infections. Over the last decade, it was discovered that TCDD further induces Tregs which are beneficial in fighting autoimmune diseases such as type I diabetes [30], multiple sclerosis [7] and colitis [31], and also in reducing the severity of graft versus host disease [27]. Doubts about the impact of TCDD on Tregs have been raised, as it has been postulated that an increased number of Tregs could result from an indirect toxic effect of TCDD. It has been proposed that TCDD leads to the death of conventional T cells, while Tregs are believed to be more resistant to apoptosis [32] (reviewed in [33]). Although the subject is still debated, this hypothesis has never been proven experimentally. Indeed, TCDD does not alter the initial expansion of activated CD4+T cells in the spleen of antigen-challenged mice but promotes a premature decline in their number before effector cell development. Furthermore, TCDD did not show a direct toxic effect when tested on multiple cell lines [34]. On the other hand, accumulating evidence indicates that AhR signalling has a specific impact on Treg’s generation. First, naïve T cells isolated from AhR null mice inefficiently generated Tregs in vitro [35]. Secondly, independent groups have shown that natural AhR ligands such as the dietary AhR ligands derived from indirubin, indole-3-carbinol and indirubin-3′-oxime [36] as well as the endogenous AhR ligands ITE and Kyn promote the differentiation of Tregs that suppress autoimmunity [26, 37].
The molecular basis by which AhR signalling modulates Treg’s biology is beginning to be understood. AhR signalling directly shapes Treg differentiation by dictating the state of Foxp3 promoter methylation (fig. 1). DNA methylation in a gene promoter region is associated with loss of that gene's expression. TCDD mediates partial demethylation of Foxp3 promoter and hence enhances Foxp3 expression, while it mediates methylation of Il17 promoter and decreases the expression of IL-17, a pro-inflammatory cytokine secreted by TH17 cells [31].
Furthermore, AhR induces tolerogenic DCs that promote the generation of Tregs. Different mechanisms by which AhR tolerise DCs have been proposed. First, AhR activation promotes the induction of IDO by binding the Ido promoter that contain putative DRE consensus sequences [38]. Accumulating evidence indicate that IDO plays a pivotal role in the induction of tolerogenic DCs (reviewed in [39]). IDO depletes tryptophan in local tissue micro-environments and generates immunoregulatory catabolites, such as Kyn. Both tryptophan starvation and the presence of Kyn promote the generation of Tregs. In vivo AhR activation with TCDD induces IDO1 and IDO-like protein IDO2 which further drive Foxp3 expression [40]. In addition, a recent publication explored the link between AhR activation by Kyn and the generation of Tregs in vitro[41]. These authors showed that IDO generated by DCs leads to an increased Kyn formation that directly promotes the generation of Tregs (fig. 1).
Generation of retinoic acid (RA), the active form of vitamin A, is another mechanism by which AhR ligand induces tolerogenic DCs that support Treg’s differentiation [26]. Treatment of DCs with the AhR ligand ITE promotes the expression of the retinal dehydrogenases Aldh1a1 that enhances RA secretion (fig. 1). Notably, RA forms a complex with the nuclear receptors, RA receptors (RARs) and retinoid X receptors (RXRs) that controls the transactivation of foxp3 promoter in coordination with other transcription factor such as MAD homolog 3 (Smad3), thereby promoting Treg differentiation.
AhR signalling not only controls mouse but also human Treg’s generation. In contrary to murine Tregs, human naïve T cells differentiated with TGF-β in vitro express Foxp3 but do not acquire suppressive properties. On the other hand, addition of an AhR ligand onto naïve T cells differentiated in the presence of TGF-β induces suppressive Foxp3+ Tregs. AhR activation does not lead to increased levels of Foxp3 expression but promotes the expression of CD39, an ectonucleotidase that hydrolyses ATP and mediates suppressive activity of Treg [8, 42]. CD39 deficiency is linked with exacerbation of autoimmune diseases in murine colitis, and human polymorphism in the CD39 gene is associated with higher susceptibility to inflammatory bowel diseases [43]. In conclusion, AhR modulates the development of Tregs both in mouse and human by multiple mechanisms.
AhR and TR1 cells
TR1 cells are an important subset among IL-10-secreting regulatory T cells and are characterised by a unique profile of cytokine production: high levels of IL-10, IL-21, some IFN-γ, low levels of IL-2 and no IL-4 (table 1) (reviewed in [17]). Interestingly, TR1 cells were first described in severe combined immuno-deficient (SCID) patients who had developed long-term tolerance to stem cell allografts, suggesting that these cells suppressed immune responses in humans [44]. Indeed, during the course of human inflammatory diseases such as multiple sclerosis, the frequency and the functionality of TR1 cells are impaired [45, 46]. Studies of TR1 cells have been difficult due to the lack of known specific lineage transcription factor or surface markers which would facilitate their tracking in vivo, and the difficulty to grow them in vitro due to their low proliferative properties. Interest in TR1 cells has recently been revived by the discovery that the cytokine IL-27 is essential for their development [47]. Exogenous and endogenous AhR ligands (TCDD, FICZ or IDE) have been shown to promote TR1 cell function as they enhance the expression of key proteins involved in their biology, which are IL-10, IL-21 [9] and Granzyme B [8]. Murine naïve T cells cultivated in the presence of AhR ligands alone are not able to convert to TR1 cells in the absence of IL-27. Indeed, IL-27 induces the expression of the transcription factors AhR and of the proto-oncogene c-Maf that enables the activation of AhR signalling. IL-27 inefficiently drives TR1 cell generation in the absence of AhR or c-Maf [9]. Upon activation AhR forms a complex with c-Maf that promotes the transactivation of il10 and il21 promoters (fig. 2) [9, 48, 49]. IL-10 and IL-21 are two essential cytokines involved in TR1 cell biology: IL-10 is crucial for the suppressive characteristics of TR1 cells and IL-21 for the expansion and maintenance of TR1 cells [48] (reviewed in [49, 50]). Interestingly, it has been recently shown that the complex c-Maf and AhR also plays a role during TH17 differentiation by controlling the expression of IL-22 [51]. Mice injected with the AhR ligand FICZ are protected against the development of colitis and have marked down-regulation of inflammatory cytokines but induction of IL-22 by TH17 cells [18]. It is noteworthy that AhR and c-Maf are expressed in TR1 cells but do not drive IL-22 expression in this cell type. This promotes the idea that while AhR and c-Maf are crucial in driving the expression of cytokines involved in the differentiation of different T cell subsets, additional yet unknown IL-27-induced triggers are critical to control the transcriptional regulation of TR1 cell.
Akin to mouse TR1 cells, AhR signalling is essential for TR1 cell differentiation in humans. Consistent with results obtained in mouse TR1 cells, the human transcription factor AhR interacts with c-Maf, ultimately resulting in an enhanced IL-10 secretion from TR1 cells [9]. While these observations are in line with the role of AhR in mouse TR1 cell differentiation, activation of human CD4+ T cells with AhR ligands in the absence of IL-27 was able to drive TR1 cell differentiation. It is noteworthy that c-Maf which is essential for mouse TR1 cell differentiation is detectable in human T cells activated without IL-27. As c-Maf expression is strictly dependent on IL-27 in mice, the observation that TCDD or FICZ alone drives the expansion of human Tr1 cells could be due to their expression of c-Maf upon sole activation. Indeed, the over-expression of c-Maf induces marginal expression of IL-10 from human CD4+ T cells, while the TCDD-driven activation of AhR combined with c-Maf over-expression leads to significant IL-10 expression [8]. IL-10 is necessary but not sufficient for the function of human TR1 cells as their capacity to suppress immune responses also relies on granzyme B expression [52, 53]. Interestingly, besides increasing IL-10 expression, AhR transactivates Gzmb promoter and drives the expression of granzyme B, which contributes to the suppressive activity of TR1 cells (fig. 2) [54]. In conclusion, akin to Foxp3+ Tregs, AhR modulates the development of TR1 cells both in mouse and human using different signalling pathways.
AhR and IL-22
While evidence generated from the studies discussed above support the contention that AhR is involved in the generation of regulatory cells, ex vivo study of T cells obtained from a man intoxicated with very high levels of TCDD did not show the expansion of Foxp3+Treg cells nor of IL-10-producing T cells [55]. Interestingly, T cells from this patient exhibited an IL-22 phenotype. As discussed previously, IL-22 is a member of the IL-10-cytokine family (reviewed in [5]) that plays a crucial role in skin and intestinal mucosa [51]. To strengthen the link between AhR and IL-22 production, it has been shown that mice injected with FICZ have enhanced IL-22 production and were protected against several murine colitis models [18]. IL-10 and IL-22 belong to the same family, suggesting that those two cytokines could be regulated by similar mechanisms [5]. New insights have been obtained in two recent studies showing that AhR signalling enhances IL-22 production by directly either binding to il22 promoter in co-ordination with the transcription factor RORγt [23] or via Notch signalling [56]. While the precise molecular mechanism behind the Notch-mediated IL-22 induction remains unknown, it has been proposed that the Notch effect could be indirect by promoting the production of endogenous ligands for AhR [56].
IL-22 expression can be associated with IL-17 and is expressed in TH17 cells [57] where AhR is expressed at a high level [7, 10]. It has been proposed that AhR signalling could promote TH17 cell development. It is important to note that TCDD or ITE injections in vivo favour the development of regulatory T cells and reduce the severity of EAE disease but treatment with FICZ aggravates the disease course of EAE. While FICZ promotes TH17 cell generation in vitro, the AhR dependent induction of TH17 cells has yet to be formally confirmed in vivo. The results of those studies have, however, led to the idea that activation of AhR by different ligands could differentially influence CD4+ T cell differentiation. However, this concept needs to be formally proven. The dosis of AhR ligands used in different studies could also be critical to the impact of AhR treatment of autoimmune diseases. Finally, the new concept that TH17 cells represent a heterogeneous population could help understanding the apparently contradictory role of FICZ on TH17 cell development. TH17 cell generated in the absence of TGF-β are highly pathogenic and do not express either AhR or IL-10, while they produce both IFN-γ and IL-17. In contrast, TH17 generated in the presence of TGF-β (TH17β) express both AhR and c-Maf, produce IL-10 and are less pathogenic [58]. Indeed, c-Maf promotes the expression of the anti-inflammatory cytokine IL-10 expression in TH17 [59] and inhibits IL-22 production [60] thereby reducing the pathogenicity of TH17 cells. The putative role of AhR signalling in different subtypes of TH17 differentiated needs to be formally addressed.
Concluding remarks
A precise balance is necessary to maintain immune surveillance but at the same time prevent the development of autoimmunity. AhR ligands have been assigned valuable immunomodulatory properties and can fine-tune the immune response. As we discussed in the first part of this review, the importance of AhR signalling in gut immunity and the availability of AhR ligands through food intake has been highlighted by numerous recent publications [2, 23–25]. AhR signalling properties could be further exploited in the clinical practice, not only in the field of infectiology but also in autoimmunity. However, several points have to be resolved before their implementation in the clinical practice. First, there is a need to design AhR ligands with safe pharmacological profiles. Indeed, as we discussed previously, the most studied AhR ligand TCDD, has valuable immunomodulatory properties but is not applicable in clinical practice because of its pharmacological properties. Furthermore, the impact of distinct AhR ligands on the immune system has to be better understood. For example, while TCDD and FICZ have opposite effects on iFoxp3+Tregs or on TH17 cells, they both promote TR1 cell or can induce IL-22 secretion depending on the experimental setting. Therefore the specificity, the pharmacology and the dose-effect of each ligand will have to be carefully assessed in different diseases models. Finally, the effect of AhR ligands needs to be cautiously monitored to avoid excessive responses. For example, the endogeneous AhR ligand Kyn has been recently shown to be secreted by human tumours via the tryptophan-2,3-dioxygenase and promote the generation of Tregs in vivo [61]. While Tregs are beneficial in preventing autoimmune diseases, they are regarded as inhibitors of anti-tumour immunity and impair the development of successful immunotherapy [62].
In conclusion, AhR ligands are promising compounds for pharmaceutical drugs. However, further studies should aim at designing a new generation of AhR ligands which could specifically target different cell types of the immune system with limited side effects. More research is required to evaluate the potential of AhR targeting for the treatment of autoimmune and inflammatory diseases in humans.
Acknowledgment: I would like to thank Professor Izui for his critical review of the manuscript.
References
1 Platzer B, Richter S, Kneidinger D, Waltenberger D, Woisetschlager M, Strobl H. Aryl hydrocarbon receptor activation inhibits in vitro differentiation of human monocytes and Langerhans dendritic cells. J Immunol. 2009;183:66–74.
2 Kiss EA, Vonarbourg C, Kopfmann S, Hobeika E, Finke D, Esser C, et al. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science. 2011;334:1561–5.
3 Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. 1986;136:2348–57.
4 Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations. Annu Rev Immunol. 2010;28:445–89.
5 Rutz S, Ouyang W. Regulation of interleukin-10 and interleukin-22 expression in T helper cells. Curr Opin Immunol. 2011;23:605–12.
6 Marshall NB, Vorachek WR, Steppan LB, Mourich DV, Kerkvliet NI. Functional characterization and gene expression analysis of CD4+ CD25+ regulatory T cells generated in mice treated with 2,3,7,8-tetrachlorodibenzo-p-dioxin. J Immunol. 2008;181:2382–91.
7 Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, et al. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature. 2008;453:65–71.
8 Gandhi R, Kumar D, Burns EJ, Nadeau M, Dake B, Laroni A, et al. Activation of the aryl hydrocarbon receptor induces human type 1 regulatory T cell-like and Foxp3(+) regulatory T cells. Nat Immunol. 2010;11:846–53.
9 Apetoh L, Quintana FJ, Pot C, Joller N, Xiao S, Kumar D, et al. The aryl hydrocarbon receptor interacts with c-Maf to promote the differentiation of type 1 regulatory T cells induced by IL-27. Nat Immunol. 2010;11:854–61.
10 Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, et al. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature. 2008;453:106–9.
11 Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–6.
12 Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–61.
13 Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. 2001;27:68–73.
14 Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet. 2001;27:18–20.
15 Horwitz DA, Zheng SG, Gray JD. Natural and TGF-beta-induced Foxp3(+)CD4(+) CD25(+) regulatory T cells are not mirror images of each other. Trends Immunol. 2008;29:429–35.
16 Weiner HL. Induction and mechanism of action of transforming growth factor-beta-secreting Th3 regulatory cells. Immunol Rev. 2001;182:207–14.
17 Roncarolo MG, Gregori S, Battaglia M, Bacchetta R, Fleischhauer K, Levings MK. Interleukin-10-secreting type 1 regulatory T cells in rodents and humans. Immunol Rev. 2006;212:28–50.
18 Monteleone I, Rizzo A, Sarra M, Sica G, Sileri P, Biancone L, et al. Aryl hydrocarbon receptor-induced signals up-regulate IL-22 production and inhibit inflammation in the gastrointestinal tract. Gastroenterology. 2011;141:237–48, 248 e231.
19 Zhang L, Ma J, Takeuchi M, Usui Y, Hattori T, Okunuki Y, et al. Suppression of experimental autoimmune uveoretinitis by inducing differentiation of regulatory T cells via activation of aryl hydrocarbon receptor. Invest Ophthalmol Vis Sci. 2010;51:2109–17.
20 Marshall NB, Kerkvliet NI. Dioxin and immune regulation: emerging role of aryl hydrocarbon receptor in the generation of regulatory T cells. Ann N Y Acad Sci.1183:25–37.
21 Wang L, Zhou GB, Liu P, Song JH, Liang Y, Yan XJ, et al. Dissection of mechanisms of Chinese medicinal formula Realgar-Indigo naturalis as an effective treatment for promyelocytic leukemia. Proc Natl Acad Sci U S A. 2008;105:4826–31.
22 Adachi J, Mori Y, Matsui S, Takigami H, Fujino J, Kitagawa H, et al. Indirubin and indigo are potent aryl hydrocarbon receptor ligands present in human urine. J Biol Chem. 2001;276:31475–8.
23 Qiu J, Heller JJ, Guo X, Chen ZM, Fish K, Fu YX, et al. The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity. 2012;36:92–104.
24 Lee JS, Cella M, McDonald KG, Garlanda C, Kennedy GD, Nukaya M, et al. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat Immunol. 2011;13:144–51.
25 Li Y, Innocentin S, Withers DR, Roberts NA, Gallagher AR, Grigorieva EF, et al. Exogenous stimuli maintain intraepithelial lymphocytes via aryl hydrocarbon receptor activation. Cell. 2011;147:629–40.
26 Quintana FJ, Murugaiyan G, Farez MF, Mitsdoerffer M, Tukpah AM, Burns EJ, et al. An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2011;107:20768–73.
27 Funatake CJ, Marshall NB, Steppan LB, Mourich DV, Kerkvliet NI. Cutting edge: activation of the aryl hydrocarbon receptor by 2,3,7,8-tetrachlorodibenzo-p-dioxin generates a population of CD4+ CD25+ cells with characteristics of regulatory T cells. J Immunol. 2005;175:4184–8.
28 Knutson JC, Poland A. Response of murine epidermis to 2,3,7,8-tetrachlorodibenzo-p-dioxin: interaction of the ah and hr loci. Cell. 1982;30:225–34.
29 Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–86.
30 Kerkvliet NI, Steppan LB, Vorachek W, Oda S, Farrer D, Wong CP, et al. Activation of aryl hydrocarbon receptor by TCDD prevents diabetes in NOD mice and increases Foxp3+ T cells in pancreatic lymph nodes. Immunotherapy. 2009;1:539–47.
31 Singh NP, Singh UP, Singh B, Price RL, Nagarkatti M, Nagarkatti PS. Activation of aryl hydrocarbon receptor (AhR) leads to reciprocal epigenetic regulation of FoxP3 and IL-17 expression and amelioration of experimental colitis. PLoS One. 2011;6:e23522.
32 Taylor SR, Alexander DR, Cooper JC, Higgins CF, Elliott JI. Regulatory T cells are resistant to apoptosis via TCR but not P2X7. J Immunol. 2007;178:3474–82.
33 Stockinger B, Hirota K, Duarte J, Veldhoen M. External influences on the immune system via activation of the aryl hydrocarbon receptor. Semin Immunol. 2011;23:99–105.
34 Knutson JC, Poland A. 2,3,7,8-Tetrachlorodibenzo-p-dioxin: failure to demonstrate toxicity in twenty-three cultured cell types. Toxicol Appl Pharmacol. 1980;54:377–83.
35 Kimura A, Naka T, Nohara K, Fujii-Kuriyama Y, Kishimoto T. Aryl hydrocarbon receptor regulates Stat1 activation and participates in the development of Th17 cells. Proc Natl Acad Sci U S A. 2008;105:9721–6.
36 Benson JM, Shepherd DM. Dietary ligands of the aryl hydrocarbon receptor induce anti-inflammatory and immunoregulatory effects on murine dendritic cells. Toxicol Sci. 2011;124:327–38.
37 Nguyen NT, Kimura A, Nakahama T, Chinen I, Masuda K, Nohara K, et al. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc Natl Acad Sci U S A. 2010;107:19961–6.
38 Puccetti P, Grohmann U. IDO and regulatory T cells: a role for reverse signalling and non-canonical NF-kappaB activation. Nature reviews. 2007;7:817–23.
39 Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nature reviews. 2004;4:762–74.
40 Vogel CF, Goth SR, Dong B, Pessah IN, Matsumura F. Aryl hydrocarbon receptor signaling mediates expression of indoleamine 2,3-dioxygenase. Biochem Biophys Res Commun. 2008;375:331–5.
41 Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol. 2010;185:3190–8.
42 Borsellino G, Kleinewietfeld M, Di Mitri D, Sternjak A, Diamantini A, Giometto R, et al. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood. 2007;110:1225–32.
43 Friedman DJ, Kunzli BM, YI AR, Sevigny J, Berberat PO, Enjyoji K, et al. From the Cover: CD39 deletion exacerbates experimental murine colitis and human polymorphisms increase susceptibility to inflammatory bowel disease. Proc Natl Acad Sci U S A. 2009;106:16788–93.
44 Bacchetta R, Bigler M, Touraine JL, Parkman R, Tovo PA, Abrams J, et al. High levels of interleukin 10 production in vivo are associated with tolerance in SCID patients transplanted with HLA mismatched hematopoietic stem cells. J Exp Med. 1994;179:493–502.
45 Astier AL, Meiffren G, Freeman S, Hafler DA. Alterations in CD46-mediated Tr1 regulatory T cells in patients with multiple sclerosis. J Clin Invest. 2006;116:3252–7.
46 Martinez-Forero I, Garcia-Munoz R, Martinez-Pasamar S, Inoges S, Lopez-Diaz de Cerio A, Palacios R, et al. IL-10 suppressor activity and ex vivo Tr1 cell function are impaired in multiple sclerosis. Eur J Immunol. 2008;38:576–86.
47 Awasthi A, Carrier Y, Peron JP, Bettelli E, Kamanaka M, Flavell RA, et al. A dominant function for interleukin 27 in generating interleukin 10-producing anti-inflammatory T cells. Nat Immunol. 2007;8:1380–9.
48 Pot C, Jin H, Awasthi A, Liu SM, Lai CY, Madan R, et al. Cutting edge: IL-27 induces the transcription factor c-Maf, cytokine IL-21, and the costimulatory receptor ICOS that coordinately act together to promote differentiation of IL-10-producing Tr1 cells. J Immunol. 2009;183:797–801.
49 Pot C, Apetoh L, Awasthi A, Kuchroo VK. Molecular pathways in the induction of interleukin-27-driven regulatory type 1 cells. J Interferon Cytokine Res. 2010;30:381–8.
50 Pot C, Apetoh L, Kuchroo VK. Type 1 regulatory T cells (Tr1) in autoimmunity. Semin Immunol. 2011;23:202–8.
51 Sonnenberg GF, Fouser LA, Artis D. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat Immunol. 2011;12:383–90.
52 Grossman WJ, Verbsky JW, Barchet W, Colonna M, Atkinson JP, Ley TJ. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity. 2004;21:589–601.
53 Kemper C, Chan AC, Green JM, Brett KA, Murphy KM, Atkinson JP. Activation of human CD4+ cells with CD3 and CD46 induces a T-regulatory cell 1 phenotype. Nature. 2003;421:388–92.
54 Magnani CF, Alberigo G, Bacchetta R, Serafini G, Andreani M, Roncarolo MG, et al. Killing of myeloid APCs via HLA class I, CD2 and CD226 defines a novel mechanism of suppression by human Tr1 cells. Eur J Immunol. 2011;41:1652–62.
55 Brembilla NC, Ramirez JM, Chicheportiche R, Sorg O, Saurat JH, Chizzolini C. In vivo dioxin favors interleukin-22 production by human CD4+ T cells in an aryl hydrocarbon receptor (AhR)-dependent manner. PLoS One. 2011;6:e18741.
56 Alam MS, Maekawa Y, Kitamura A, Tanigaki K, Yoshimoto T, Kishihara K, et al. Notch signaling drives IL-22 secretion in CD4+ T cells by stimulating the aryl hydrocarbon receptor. Proc Natl Acad Sci U S A. 2011;107:5943–8.
57 Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, et al. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med. 2006;203:2271–9.
58 Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, et al. Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature. 2010;467:967–71.
59 Xu J, Yang Y, Qiu G, Lal G, Wu Z, Levy DE, et al. c-Maf regulates IL-10 expression during Th17 polarization. J Immunol. 2009;182:6226–36.
60 Rutz S, Noubade R, Eidenschenk C, Ota N, Zeng W, Zheng Y, et al. Transcription factor c-Maf mediates the TGF-beta-dependent suppression of IL-22 production in T(H)17 cells. Nat Immunol. 2011;12:1238–45.
61 Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. 2011;478:197–203.
62 Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nature reviews. 2006;6:295–307.