AA amyloidosis: basic knowledge, unmet needs and future treatments
DOI: https://doi.org/10.4414/smw.2012.13580
Laura
Obici, Giampaolo
Merlini
Summary
Systemic AA amyloidosis is a long-term complication of several chronic inflammatory disorders, including rheumatoid arthritis, ankylosing spondylitis, autoinflammatory syndromes, Crohn’s disease, malignancies and conditions predisposing to recurrent infections. Organ damage results from the extracellular deposition of proteolytic fragments of the acute-phase reactant serum amyloid A (SAA) as amyloid fibrils. A sustained high concentration of SAA is the prerequisite for developing AA amyloidosis. However, only a minority of patients with long-standing inflammation actually presents with this complication, pointing to the existence of disease-modifying factors, the best characterised of which being SAA1 genotype. The kidneys, liver and spleen are the main target organs of AA amyloid deposits. In more than 90% of patients proteinuria, nephrotic syndrome and/or renal dysfunction dominate the clinical picture at onset. If not effectively treated, this disease invariably leads to end stage kidney disease and renal replacement therapy, that are still associated with a poor outcome.
Although the incidence of AA in rheumatoid arthritis and other chronic arthritides has continuously decreased over the past ten years, thanks to the increasing availability of more effective anti-inflammatory and immunosuppressive therapies, AA remains a life-threatening disease with several areas of uncertainty and unmet needs, deserving continuous efforts at prevention and effective treatment. The deeper understanding of the molecular mechanisms of amyloid formation and regression is now driving the development of novel treatments targeting different steps in the amyloidogenic cascade. These therapies will hopefully improve the quality of life and outcome of these patients in a near future.
Introduction
The past ten years have witnessed significant progress in the clinical management of systemic amyloidoses, thanks to the increasing availability of refined diagnostic techniques, the identification of novel prognostic markers for risk-stratification and the development of more effective agents that suppress or reduce the circulating amyloidogenic precursors [1]. Such improvements have been paralleled by a greater awareness of this group of diseases in the medical community, overall leading to earlier recognition and more tailored treatments.
A significant impact of these advances has been seen in systemic, reactive AA amyloidosis, a long-recognised, severe complication of several chronic inflammatory diseases. Frequent and well-established conditions associated with AA in Western countries, particularly chronic inflammatory arthritis, have dramatically benefited from novel anti-rheumatic treatments in the past fifteen years. As anticipated by Hazenberg and van Rijswijk in 2000 [2], a decline in the actual incidence of AA amyloidosis complicating rheumatoid arthritis (RA) could be foreseen to occur with a lag phase of at least 10 years from intensification of treatment.
Such reduction in the occurrence of AA in RA and other arthritides is now established [3] and is also reflected by a change in the relative frequency of the inflammatory diseases underlying AA in patients diagnosed over the past ten years at some centres [4, 5].
However, once it develops, AA amyloidosis continues to challenge our treatment capabilities, because of the lack of therapies that adequately suppress inflammation in several cases and the frailty of these patients when renal function significantly declines. Overall this remains a complex and potentially life-threatening disease with several areas of uncertainty and unmet needs, deserving continuous efforts to prevent and treat it effectively.
In this review we discuss current standards in the diagnosis and treatment of AA amyloidosis, highlighting areas of uncertainty and foreseeing further improvements related to the expanding understanding of the molecular mechanisms of amyloid formation and regression.
Molecular mechanisms of AA amyloidosis
The conversion of the circulating soluble protein serum amyloid A (SAA) into stable, highly ordered amyloid fibrils that accumulate extracellularly causing organ damage is a multi-step process that is primed by a persistently and abnormally high concentration of this protein in plasma. As an acute phase reactant secreted by the liver under the transcriptional control of interleukin-1 (IL-1) and IL-6, SAA increases up to 1000 fold following an inflammatory stimulation. If such stimuli persist, as occurs in several chronic diseases, SAA concentration may reach a critical threshold over which it becomes prone to aggregation. This property is common to other soluble proteins that may undergo misfolding, aggregation and ultimately generation of cross-β-sheet amyloid fibrils in vivo following an absolute or relative increase in their concentration in a tissue or compartment [6]. Overall, at least 25 different proteins are known to be able to form disease-associated, systemic or localised amyloid deposits in humans [1]. The first step of the fibrillation process is the partial unfolding of the amyloidogenic precursor to soluble, monomeric, non-native conformations that self-assemble into oligomeric aggregates characterised by a very fast kinetic of association and dissociation, highly sensitive to the interaction with hydrophobic surfaces, pH, ion strength and other factors that slightly fluctuate in the extracellular space [6]. Based on these highly dynamic conformational changes, structurally different soluble oligomers can arise at the same time, not all equally harmful, with only a few ultimately forming amyloid fibrils [7]. In addition, some of these pre-amyloid oligomers are well known to exert a direct cytotoxic effect [8]. Increasing evidence also supports the possibility that multiple, morphologically distinct forms of amyloid fibrils may be generated from a single amyloidogenic protein through different aggregation pathways [7].
Together with the primary amino acid structure of the protein itself, cellular and extracellular factors such as proteolytic remodelling and interaction with matrix components are known to impact on these aggregation pathways [6]. In AA amyloidogenesis, in which fibrils are invariably formed by N-terminal fragments spanning the first 66-76 amino acids of SAA, a pivotal role of proteolytic remodelling is supported by several studies [9, 10], although it has not yet been ascertained whether this event takes place before or after the protein starts to aggregate. Interactions with glycosaminoglycans are also known to promote misfolding and aggregation of SAA both in vitro and in vivo and are able to accelerate it by acting as a scaffold for polymerisation [11, 12].
Overall, fibrillation is an unfavourable process in which proteins must overcome a thermodynamic and kinetic barrier and therefore is not unexpected that it occurs over a long time. However, once a first nucleus is formed, it acts as a seed for further polymerisation as the kinetic barrier collapses resulting in the accelerated growth of amyloid deposits. Such a nucleation-elongation model occurs for most amyloidogenic proteins. Similarly to what was originally proposed for prions, the structural determinants that drive fibril growth through the selection of additional molecules with similar conformation have now been increasingly characterised for a variety of other proteins and peptides [13]. These mechanisms of specific recognition among proteins are also at the basis of the ability of some molecules to cross-seed amyloid of different species. This is not only observed experimentally but it has also been demonstrated to occur in humans, as recently reported by Larsson et al, who showed that arterial wall deposits formed by medin can cross-seed AA amyloid fibrils [14].
These molecular recognition events also account for the potential infectivity of amyloid seeds, i.e., the well-known ability to experimentally induce amyloidosis in animals by inoculating even tiny amounts of a template of pre-formed fibrils. As for prions, AA seeding has been supported to occur also in vivo, by means of oro-faecal spreading of amyloid-contaminated faeces in captive cheetah [15]. Whether transmission of AA amyloid particles among species, including humans, could also take place through the food chain is still a matter of speculation [16].
The pathogenic relevance of seeding as a general mechanism underlying amyloid diseases is also supported by increasing evidence that in animals infected with Aβ, tau or other proteins associated with neurodegenerative diseases, early aggregates are able to spread through neuronal pathways and other still unknown transport mechanisms within the body, to reach the areas where the final damage occurs [17]. In mice, monocytes have been shown to serve as vehicles for AA amyloid seeds [18]. Together with the well-known observation that the spleen is the first organ where AA amyloidosis develops in mice, it is tempting to speculate that such seeding mechanisms might be involved not only in transmission and acceleration of amyloidogenesis but could also contribute to disease dissemination to target organs, resulting in seed-related tissue specificity [16, 17].
Old and novel diseases causing AA
The spectrum of inflammatory diseases associated with AA has slowly but continuously changed over time. Chronic infections characterised by significant acute phase reaction, such as tuberculosis and osteomyelitis, have long been the main causes of AA amyloidosis and still remain relevant in some areas in developing countries [19]. In the past decades, chronic arthritis, particularly rheumatoid arthritis, ankylosing spondylitis and juvenile idiopathic arthritis have become major causes of AA amyloidosis, with RA being responsible for 30–60% of patients in different series [20]. However, thanks to more aggressive treatment schedules and to the increasing availability of anti-TNF treatments, the incidence of AA in chronic arthritides has slowly decreased in the past ten years [3–5]. This has led to a relative increase in the rate of other conditions that are well-recognised to significantly associate with AA, such as Crohn’s disease, hereditary periodic fevers, malignancies, systemic vasculitides and diseases predisposing to recurrent infections, including cystic fibrosis, bronchiectasis, epidermolysis bullosa, cyclic neutropenia, acquired or inherited immunodeficiencies, injection-drug use and acne conglobata (table 1). In a significant subset of patients, however, a clear-cut history for a defined chronic inflammatory condition is not present at diagnosis, although inflammatory markers might have been elevated for a relatively long time, in the absence of symptoms. In some of these patients a thorough work-up may actually lead to a final diagnosis, sometimes unveiling novel conditions associated with renal AA amyloidosis [21]. Increasing significance has recently been paid to obesity [22] and hepatitis B [5] although these are mostly mild inflammatory diseases. Interestingly, in a 13- year-old boy in which AA secondary to hepatitis B developed, a N-terminal mutation in the SAA gene was reported, that could potentially contribute to amyloidogenesis [23].
Finally, in most series the underlying disease ultimately remains unknown in 5–10% of patients. The potential contribution of still uncharacterised environmental or genetic factors is likely to play a role in these cases. Recently, in a 40-year-old woman with AA and persistent inflammation of unknown aetiology we found by Comparative Genomic Hybridisation (CGH) array, a technique which allows detection of copy number variations (i.e., small deletions and duplications) in the genome, a large (4.6 Mb) deletion on the long arm of chromosome 15 (unpublished data). The pathogenic significance of this finding remains elusive. However, it is intriguing that this region hosts SELS, the gene for selenoprotein S, which is a 189 amino acid protein expressed in many tissues and containing a selenocysteine residue at its active site. Selenoprotein S is an endoplasmic reticulum (ER)-associated protein that participates in the processing and removal of misfolded proteins from ER to cytosol, for proteasome degradation. Moreover, it has been found to interact with SAA and in vitro its suppression is associated with increased release of proinflammatory cytokines [24].
Other factors largely unknown could contribute to the individual susceptibility or resistance to the amyloid deposition and particularly scrutinised is the repertoire of intracellular and extracellular chaperone that each single individual could deploy against protein misfolding and aggregation [25].
|
Table 1: Inflammatory diseases associated with AA amyloidosis. |
|
Inflammatory arthritis
Rheumatoid arthritis
Ankylosing spondylitis
Adult Still’s disease
Juvenile idiopathic arthritis
Psoriatic arthritis
Gout
Inflammatory bowel diseases
Crohn’s disease
Ulcerative colitis
Hereditary and acquired immunodeficiencies
Common variable immunodeficiency
Hypogammaglobulinaemia
X-linked agammaglobulinaemia
Cyclic neutropenia
HIV/AIDS
Others
Obesity (?)
Sarcoidosis
SAPHO sindrome
Schniztler syndrome |
Neoplastic diseases
Castleman’s disease
Hodgkin’s lymphoma
Waldenstrom’s macroglobulinaemia
Hairy cell leukaemia
Hepatic adenoma
Renal cell carcinoma
Adenocarcinoma of the lung
Adenocarcinoma of the gut
Mesothelioma
Chronic infections
Bronchiectasis
Osteomyelitis
Tuberculosis
Chronic pyelonephritis
Leprosy
Whipple’s disease
Chronic cutaneous ulcers
Hepatitis B (?) |
Hereditary autoinflammatory diseases
Familial Mediterranean fever
TNF receptor-associated periodic syndrome (TRAPS)
Muckle-Wells syndrome
NOMID/CINCA syndrome
Hyper-IgD syndrome
Systemic vasculitides
Behcet’s disease
Polyarteritis nodosa
Giant cell arteritis
Takayasu’s arteritis
Polymyalgia rheumatica
Conditions predisposing to chronic infections
Cystic fibrosis
Epidermolysis bullosa
Injected-drug use
Jejuno-ileal bypass
Paraplegia |
Is it possible to predict AA amyloidosis?
It is well known that only a minority of patients with a long-standing inflammatory disease ultimately develop AA amyloidosis. Protracted disease duration is expected to increase the risk of this complication. Median duration of inflammatory disease at diagnosis was 17 years in a large UK series [26]. However, exposure to high levels of SAA even for decades may not result in clinically overt AA. On the contrary, rapidly progressive kidney damage due to AA still occurs in children with familial Mediterranean fever before colchicine treatment is established, particularly in some populations, in which the latency period is therefore significantly shorter [27]. Several studies have focused on putative environmental or genetic factors that might increase susceptibility to AA and have prognostic significance, with the aim of identifying patients that would benefit from more aggressive treatments. To date, SAA genotype is the only established variable that significantly affects the risk of development of AA. Two different SAA isoforms, SAA1 and SAA2, account for the rise of SAA levels during the acute-phase response. In humans, three main SAA1 alleles are known, indicated as SAA1.1, SAA1.3 and SAA1.5, that differ for single amino acid substitutions at codons 52 and 57. In Japanese, homozygosity for SAA1.3 significantly increases the risk of AA and it is also associated with a shorter latency period prior to AA onset, more severe AA-related symptoms and poorer survival [28]. On the contrary, Caucasian subjects homozygous for SAA1.1develop AA three to seven times more frequently than other genotypes [25]. Several questions remain open on the biological reasons of this discrepancy among different populations and on the molecular mechanisms by which these alleles variably affect AA development [29].
Early and accurate diagnosis of AA amyloidosis
In more than 90% of cases the first sign of AA onset is glomerular proteinuria, which may exhibit an early association with slightly impaired renal function [25]. If left untreated, kidney damage invariably progresses to nephrotic syndrome and ultimately renal failure occurs, reaching end stage kidney disease (ESKD) in an unpredictable time course. Symptomatic autonomic dysfunction and cardiac amyloidosis are usually late manifestations.
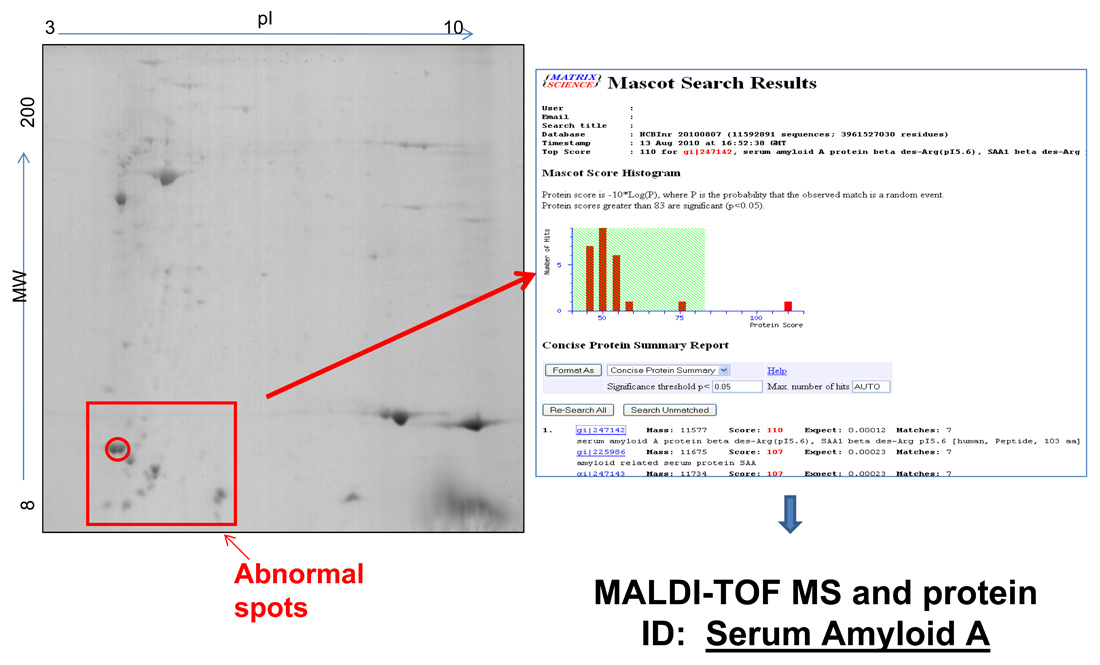
Figure 1
Typical 2D-PAGE map of fat tissue in AA amyloidosis.A 73-year old man presented with signs of renal and gastrointestinal amyloidosis. Immunohistochemistry classified the disease as ALκ type. However, due to the absence of a serum or urine monoclonal component, with normal serum free kappa light chains and κ/λ ratio, this result was considered inconclusive. Proteomic analysis by means of two-dimensional polyacrylamide gel electrophoresis (2D-PAGE, left panel) showed the presence of abnormal spots (in the box) that are not found in controls. Mass spectrometry analysis of excised spots, followed by data base search (right panel) characterized these spots as N-terminal fragments of SAA, allowing definite typing of amyloidosis.
Repeated measurements of microalbuminuria, cystatin C and estimated creatinine clearance could help identifying patients in the earlier phases of the disease [30, 31]. Screening for subclinical AA amyloid deposits is also routinely performed in some centres in patients with RA, to identify those at higher risk for clinically overt AA amyloidosis. Although it is known that subclinical AA may occur without further development of organ damage, the identification of even asymptomatic amyloid deposits should prompt to a more effective control of inflammation [32].
To screen for subclinical AA or to ascertain the diagnosis, abdominal fat needle biopsy is now thoroughly recommended as the first choice approach to search for apple-green refringent amyloid deposits on Congo red stained tissue slides examined under polarised light [33, 34]. This technique was originally developed as a non-invasive method to look for AA amyloid deposits in patients with RA [35], overcoming bleeding risks potentially associated with organ biopsy, and it has subsequently been validated by several groups [34]. This procedure is quick, safe and inexpensive and can also be performed repeatedly over time. In AA as well as in AL amyloidosis, its sensitivity and specificity are reported to be >90% and 100% respectively, in experienced hands [36]. Multiple sampling, correct staining protocol, long-observation, good polariser, room darkness are important to overcome low sensibility due the presence of scanty and unevenly distributed amyloid deposits and to avoid overdiagnosis due to white birefringence of collagen [33, 34].
Gastrointestinal biopsies are similarly suitable for the diagnosis of AA. Duodenal biopsy has higher sensitivity compared to colonic and rectum biopsies [37]. In Japan, screening by upper gastrointestinal tract biopsy is common, allowing early identification of amyloid in patients with RA [38]. A duodenal biopsy should particularly be considered in patients with chronic kidney disease in whom kidney biopsy cannot be performed, or in those with a negative abdominal fat aspirate [39].
Labial minor salivary glands are another easy and informative site to approach the diagnosis in patients with a negative abdominal fat biopsy [40]. In a recent study, its sensitivity and specificity in detecting AA amyloidosis were reported to be 86% and 100% respectively [41].
Organ biopsy, particularly kidney biopsy, is recommended in cases in which less invasive approaches have not been diagnostic and clinical suspicion remains high. With this in mind, in one retrospective series the actual incidence of bleeding complication was not higher in patients with amyloidosis compared with patients without it [42].
Typing of amyloid deposits is always mandatory. Immunohistochemistry with anti-SAA antibody on paraffin-embedded or frozen sections routinely allows a straightforward and definite diagnosis in most patients [33], provided that a panel of validated antibodies is tested with appropriate controls [43, 44]. Careful interpretation of immunohistochemistry results is recommended, particularly in patients with a coincidental monoclonal gammopathy or carrying a genetic variant in one amyloidogenic protein. Cases ultimately inconclusive by standard immunohistochemistry or immunofluorescence may occur [33, 44]. Additionally, in patients in whom the underlying inflammatory aetiology remains unknown, further characterisation of amyloid deposits by a method different from immunohistochemistry should be performed. Such methods, that are mostly available at referral centres, include immuno-electron microscopy [45], Western-Blotting [46], extraction and amino acid sequencing of amyloid proteins from tissues [47], and proteomics-based methodologies, recently reviewed by Lavatelli and Vrana [48]. A prototypic case solved by proteomic analysis of abdominal fat is reported in figure 1.
Prognosis
Once clinically overt kidney damage due to AA amyloidosis occurs, the prognosis is dictated by the effective control of the underlying inflammatory condition. In the largest series of AA patients reported to date, in which median survival from diagnosis was 133 months, it was clearly established that renal prognosis and survival significantly correlate with the SAA concentration during follow-up [26]. Even a mild raise in SAA levels (two times the upper reference limit) increases the risk of death by five fold. When the median annual concentration persists above 45 mg/L, such risk becomes more than 10-fold. On the contrary, SAA concentration within the lower reference range significantly reduces the risk of death and correlates with a regression of the amount of amyloid deposited, as assessed by SAP scintigraphy. Recently, in another series of AA patients with chronic arthritides, proteinuria at baseline was found to predict mortality [49], which is consistent with poor prognosis associated with reduced serum albumin in other studies [26].
End-stage kidney disease requiring renal replacement therapy is associated with bad outcome. In a series of Finnish patients the median survival times on renal replacement therapy, either haemodialysis or peritoneal dialysis, were 2.11 years for RA, 2.37 years for ankylosing spondylitis and 3.05 for juvenile idiopathic arthritis, with 5-year survival rate being 18% in RA [50]. These figures are in agreement with data reported in Italian, Japanese and other populations [26, 51–52].
Major causes of death in patients with ESKD are heart failure, including a higher rate of sudden death after haemodialysis is started, gastrointestinal bleeding and perforation, and an increased frequency of fatal infections [52].
Goals of therapy
A rapid and complete control of the inflammatory process is the main goal of treatment in patients with AA amyloidosis, and thus basically depends on the nature of the underlying condition. SAA concentration should be strictly monitored over time to assess biochemical response to treatment. Persistence of asymptomatic inflammation is frequent and treatment should aim at suppressing it as much as possible. In particular, the risk of recurrent inflammatory flare-ups should never be overlooked. If a novel boost of inflammation arises, even when an adequate SAA control was obtained for a sufficient period of time to allow recovery of kidney damage, sudden worsening of proteinuria and/or rapid deterioration of renal failure still may occur.
Whether this is due to the accelerated growth of deposits on pre-existing fibrils or might be related to a possible direct cytotoxic effect of pre-fibrillar oligomeric aggregates of SAA, still remains a matter of debate. However, the rapidity by which such worsening occurs and can be promptly reversed by a quick control of inflammation is reminiscent of the strict relationship between serum free light chains and cardiac toxicity in AL amyloidosis, in which the role of cardiotoxic aggregates has been advocated [53].
Recently, the importance of a tight control of blood pressure to improve renal response has also been highlighted, to prevent glomerular haemodynamic changes associated with suboptimal control of hypertension [54].
Early intervention with anti-TNFα agents, with or without methotrexate, is increasingly recommended in patients with AA secondary to rheumatoid arthritis. However, evidence still relies on small case series with a relatively short follow-up, and these do not yet allow a comparison between different treatment schedules. An excellent review of these studies has just been published [55]. In addition, a recent prospective, controlled study supports long-term efficacy of anti-TNF treatment in AA associated with rheumatic diseases, even in spite of suboptimal suppression of inflammatory markers. In this series renal dysfunction improved in 54% of patients and stabilised in 16%. However, a higher frequency of infections, including fatal sepsis, was observed in AA patients compared to controls, so the actual survival benefit still remains undetermined [49].
In autoinflammatory diseases, including familial Mediterranean fever (FMF), a dramatic change in our treatment capabilities occurred in the past ten years following the discovery that most of these pathologies are driven by abnormal IL-1β secretion and can be better controlled by means of anti-IL agents [56]. Even in FMF, which remains effectively treated by daily colchicine in more than 95% cases, anti IL-1 treatment may play a significant role in patients unresponsive or poorly tolerant to this therapy at standard doses, particularly when ESKD occurs [57].
Several areas of uncertainty still remain in the management of AA amyloidosis. One relevant issue is how aggressive the treatment should become in patients that show evidence of subclinical amyloid deposits. As only a minority of these patients probably will end up with kidney damage, the question is how to balance the potential benefits of preventing this complication towards the increased risks, particularly infective, related to immunosuppressive and anti-cytokine agents. A close follow-up with repeated SAA evaluations is mandatory. Although no prospective studies are available, SAA levels persistently over the threshold of 10 mg/L combined with at risk genotype should prompt to a more strict control of the inflammation.
Finally, renal transplantation is an important option in patients with AA in which a stable control of the underlying disease has been achieved. However, appropriate patient selection is strongly recommended due to a significantly higher incidence of heart failure and infections in AA individuals, in whom 5- and 10-year mortality remains higher compared to controls, although graft survival does not differ [58].
Emerging treatments
As discussed, once organ damage develops, the outcome of patients with AA can be poor, either because of lack or limited response to treatment, severe adverse events related to anti-inflammatory drugs or due to the absence of long-term effective therapies, i.e. in patients with cystic fibrosis and bronchiectasis. There is therefore an urgent need for additional treatment options that might act on different steps of the amyloidogenic cascade. Some novel drugs are now in the pipeline. One approach is the inhibition of the interaction of SAA with matrix glycosaminoglycans, which are known to promote protein misfolding and aggregation. In a phase II/III placebo-controlled study with eprodisate, a negatively charged sulfonated molecule, a significant reduction in renal function deterioration was observed in treated patients compared to controls, independently from the circulating SAA levels [59]. These promising results await further confirmation from an on-going phase III trial ( http://www.clinicaltrial.gov NCT01215747).
Another strategy steps from seminal studies on the pathogenic role of the common constituent of all amyloid deposits, serum amyloid P component (SAP), which binds amyloid fibrils and protect them from proteolysis and clearance. To promote fibril re-absorption, a high-affinity ligand of SAP, the bis-D-proline compound CPHPC was first developed [60]. Treatment with CPHPC depletes circulating levels of SAP by more than 90% but some SAP remains bound to fibrils. Recently however, Bodin et al. demonstrated that immunotherapy with anti-SAP antibody is able to eliminate visceral amyloid deposits in mice previously exposed to CPHPC [61].
Finally, a novel approach is represented by antisense oligonucleotides targeted to suppress the production of SAA. Preliminary data support effective reduction of SAA levels by this strategy, coupled with a limitation in AA deposition in mice [62].
Conclusions
Early diagnosis and rapid control of the underlying inflammatory disease are of utmost importance to prevent irreversible organ damage and to improve survival in patients with AA amyloidosis. Monitoring of patients with sustained inflammatory diseases by serial evaluations of SAA, CRP, microalbuminuria, proteinuria, serum cystatin C and eGFR, combined with a periodic search for subclinical amyloid deposits on abdominal fat aspirate or duodenal biopsy, might help in identifying patients at higher risk of developing AA. Additionally, the assessment of SAA1 genotype may also contribute to guide treatment approach. As for other systemic amyloidoses, novel treatment approaches for AA are under development. The results of the on-going trial with eprodisate are eagerly awaited to confirm its efficacy in preventing renal deterioration in such fragile patients.
References
1 Merlini G, Seldin DC, Gertz MA. Amyloidosis: pathogenesis and new therapeutic options. J Clin Oncol. 2011;29:1924–33.
2 Hazenberg B, Van Rijswijk MH. Where has secondary amyloid gone? Ann Rheum Dis. 2000;59:577–79.
3 Immonen K, Finne P, Gronhagen-Riska C, Petterson T, Klaukka T, Kautiainen H, et al. A marked decline in the incidence of renal replacement therapy for amyloidosis associated with inflammatory rheumatic diseases – data from nationwide registries in Finland. Amyloid. 2011;18:25–8.
4 Hunter J, McGregor L. Do inflammatory rheumatic diseases still cause as much harm through amyloidosis? Amyloid. 2011;18 (Suppl 1):208–10.
5 Girnius S, Dember L, Doros G, Skinner M. The changing face of AA amyloidosis: a single center experience. Amyloid. 2011;18 (Suppl 1):226–8.
6 Bellotti V, Chiti F. Amyloidogenesis in its biological environment: challenging a fundamental issue in protein misfolding diseases. Curr Opin Struc Biol. 2008;18:771–9.
7 Uversky VN. Mysterious oligomerization of the amyloidogenic proteins. FEBS J. 2010;277:2940–53.
8 Sakono M, Zako T. Amyloid oligomers: formation and toxicity of Abeta oligomers. FEBS J. 2010;277:1348–58.
9 Stix B, Kahne T, Sletten K, Raynes J, Roessner A, Rocken C. Proteolysis of AA amyloid fibril proteins by matrix metalloproteases-1 -2, and 3. Am J Pathol. 2001;159:561–70.
10 van der Hilst JCH, Yamada T, op den Camp HJM, van der Meer JWM, Drenth JPH, Simon A. Increased susceptibility of serum amyloid A 1.1 to degradation by MMP-1: potential explanation for higher risk of type AA amyloidosis. Rheumatology. 2008;47:1651–4.
11 Elimova E, Kisilevsky R, Szarek WA, Ancsin JB. Amyloidogenesis recapitulated in cell culture: a peptide inhibitor provides direct evidence for the role of heparan sulfate and suggests a new treatment strategy. FASEB J. 2004;18:1749–51.
12 Li JP, Galvis ML, Gong F, Zhang X, Zcharia E, Metzger S, et al. In vivo fragmentation of heparan sulphate by heparanase overexpression renders mice resistant to amyloid protein A amyloidosis. Proc Natl Acad Sci USA 2005;102:6473–7.
13 Ma B, Nussinov R. Selective molecular recognition in amyloid growth and transmission and cross-species barriers. J Mol Biol. 2011; doi:10.1016/j.jmb.2011.11.023.
14 Larsson A, Malmstrom S, Westermark P. Signs of cross-seeding: aortic medin as a trigger for protein AA deposition. Amyloid. 2011;18:229–34.
15 Zhang B, Une Y, Fu X, Yan J, Ge F, Yao J, et al. Fecal transmission of AA amyloidosis in the cheetah contributes to high incidence of disease. Proc Natl Acad Sci USA 2008;105:7263–8.
16 Westermark GT, Westermark P. Serum amyloid A and protein AA: Molecular mechanisms of a transmissible amyloidosis. FEBS Lett. 2009;583:2685–90.
17 Jucker M, Walker LC. Pathogenic protein seeding in Alzheimer disease and other neurodegenerative disorders. Ann Neurol. 2011;70:532–40.
18 Sporanova J, Nystrom SN, Westermark GT. AA-amyloidosis can be transferred by peripheral blood monocytes. PLoS One 2008;3:e3308.
19 Dixit R, Gupta R, Dave L, Prasad N, Sharma S. Clinical profile of patients having pulmonary tuberculosis and renal amyloidosis. Lung India. 2009;26:41–5.
20 Stankovic C, Grateau G. Is there any treatment for inflammatory amyloidosis? Joint Bone Spine. 2011;78:7–9.
21 Toledo K, Perez MJ, Espinosa M, Ortega R, Aljama P. Antisynthetase syndrome without myositis secondary to AA amyloidosis: a non-described association. Nefrologia. 2011;31:107–27.
22 Alsina E, Martin M, Panades M, Fernandez E. Renal AA amyloidosis secondary to morbid obesity? Clin Nephrol. 2009;72:312–4.
23 Saha A, Theis JD, Vrana JA, Dubey NK, Batra VV, Sethi S. AA amyloidosis associated with hepatitis B. Nephrol Dial Transplant. 2011;26:2407–12.
24 Curran JE, Jowett JB, Elliott KS, Gao Y, Gluschenko K, Wang J, et al. Genetic variation in selenoprotein S influences inflammatory response. Nat Genet. 2005;37:1234–41.
25 van der Hilst JCH. Recent insights into the pathogenesis of type AA amyloidosis. ScientificWorldJournal. 2011;11:641–50.
26 Lachmann HJ, Goodman HJB, Gilbertson JA, Gallimore JR, Sabin CA, Gillmore JD, et al. Natural history and outcome in systemic AA amyloidosis. N Engl J Med. 2007;356:2361–71.
27 Bilginer Y, Akpolat T, Ozen S. Renal amyloidosis in children. Pediatr Nephrol. 2011;26:1215–2.
28 Nakamura T, Higashi S, Tomoda K, Tsukano M, Baba S, Shono M. Significance of SAA1.3 allele genotype in Japanese patients with amyloidosis secondary to rheumatoid arthritis. Rheumatology. 2006;45:43–9.
29 Obici L, Raimondi S, Lavatelli F, Bellotti V, Merlini G. Susceptibility to AA amyloidosis in rheumatic diseases: a critical overview. Arthritis Rheum. 2009;61:1435–40.
30 Sato H, Kuroda T, Tanabe N, Ajiro J, Wada Y, Murakami S, et al. Cystatin C is a sensitive marker for detecting a reduced glomerular filtration rate when assessing chronic kidney disease in patients with rheumatoid arthritis and secondary amyloidosis. Scand J Rheumatol. 2010;39:33–7.
31 Tishko AN, Lapin SV, Vavilova TV, Totolian AA. Early diagnostics of kidney damage in longstanding rheumatoid arthritis and amyloidosis. Amyloid. 2011;18(Suppl 1):217–20.
32 Immonen K, Helin H, Lehtinen K, Hakala M. The usefulness of subcutaneous fat tissue aspiration biopsy for early confirmation of amyloidosis in patients with active ankylosing spondylitis: comment on the article of van Gameren et al. Arthritis Rheum. 2007;56:2467–8.
33 Picken MM. Amyloidosis-Where are we now and where are we heading? Arch Pathol Lab Med. 2010;134:545–51.
34 Westermark P. Amyloid diagnosis, subcutaneous adipose tissue, immunohistochemistry and mass spectrometry. Amyloid. 2011;18:175–6.
35 Westermark P, Stenqvist B. A new method for the diagnosis of systemic amyloidosis. Arch Intern Med. 1973;132:522–3.
36 Van Gameren II, Hazenberg BPC, Bijzet J, van Rijswijk MH. Diagnostic accuracy of subcutaneous abdominal fat tissue aspiration for detecting systemic amyloidosis and its utility in clinical practice. Arthritis Rheum. 2006;54:2015–21.
37 Miyaoka M, Matsui T, Hisabe T, Yano Y, Hirai F, Takaki Y, et al. Clinical and endoscopic features of amyloidosis secondary to Crohn’s disease: diagnostic value of duodenal observation and biopsy. Dig Endosc. 2011;23:157–65.
38 Kuroda T, Tanabe N, Kobayashi D, Wada Y, Murakami S, Nakano M, et al. Significant association between renal function and area of amyloid deposition in kidney biopsy specimens in reactive amyloidosis associated with rheumatoid arthritis. Rheumatol Int 2011 e-pub.
39 Yilmaz M, Unsal A, Sokmen M, Harmankaya O, Alkim C, Kabukcuoglu F, et al. Duodenal biopsy for diagnosis of renal involvement in amyloidosis. Clin Nephrol. 2012;77:114–8.
40 Foli A, Palladini G, Caporali R, Verga L, Morbini P, Obici L, et al. The role of minor salivary gland biopsy in the diagnosis of systemic amyloidosis: results of a prospective study in 62 patients. Amyloid. 2011;18(Suppl 1):80–2.
41 Sacsaquispe SJ, Antunez-de Mayolo EA, Vicetti R, Delgado WA. Detection of AA-type amyloid protein in labial salivary glands. Med Oral Pat Oral Cir Bucal. 2011;16:e149–52.
42 Soares SM, Fervenza FC, Lager DJ, Gertz MA, Cosio FG, Leung N. Bleeding complications after transcutaneous kidney biopsy in patients with systemic amyloidosis: single-center experience in 101 patients. Am J Kidney Dis. 2008;52:1079–83.
43 Linke RP, Oos R, Wiegel NM, Nathrath WB. Classification of amyloidosis: Misdiagnosis by way of incomplete immunohistochemistry and how to prevent it. Acta Histochem. 2006;108:197–208.
44 Schonland S, Hegenbart U, Bochtler T, Mangatter A, Hansberg M, Ho AD, et al. Immunohistochemistry in the classification of systemic forms of amyloidosis: a systematic investigation of 117 patients. Blood. 2012;119:488–93.
45 Arbustini E, Verga L, Concardi M, Palladini G, Obici L, Merlini G. Electron and immuno-electron microscopy of abdominal fat identifies and characterizes amyloid fibrils in suspected cardiac amyloidosis. Amyloid. 2002;9:108–14.
46 Westermark P, Davey E, Lindbom K, Enqvist S. Subcutaneous fat tissue for diagnosis and studies of systemic amyloidosis. Acta Histochem. 2006;108:209–13.
47 Murphy CL, Wang S, Williams T, Weiss DT, Solomon A. Characterization of systemic amyloid deposits by mass spectrometry. Methods Enzymol. 2006;412:48–62.
48 Lavatelli F, Vrana JA. Proteomic typing of amyloid deposits in systemic amyloidoses. Amyloid. 2011;18:177–82.
49 Fernandez-Nebro A, Olivé A, Castro MC, Herranz Varela A, Riera E, Irigoyen MV, et al. Long-term TNF-α blockade in patients with amyloid A amyloidosis complicating rheumatic diseases. Am J Med. 2010;123:454–61.
50 Immonen K, Finne P, Hakala M, Kautiainen H, Pettersson T, Gronhagen-Riska C. No improvement in survival of patients with amyloidosis associated with inflammatory rheumatic diseases – data from the Finnish national registry for kidney diseases. J Rheum. 2008;35:1334–8.
51 Bergesio F, Ciciani AM, Manganaro M, Palladini G, Santostefano M, Brugnano R, et al. Renal involvement in systemic amyloidosis: an Italian collaborative study on survival and renal outcome. Nephrol Dial Transpl. 2008;23:941–51.
52 Kuroda T, Tanabe N, Sato H, Ajiro J, Wada Y, Murakami S, et al. Outcome of patients with reactive amyloidosis associated with rheumatoid arthritis in dialysis treatment. Rheumatol Int. 2006;26:1147–53.
53 Palladini G, Lavatelli F, Russo P, Perlini S, Perfetti V, Bosoni T, et al. Circulating amyloidogenic free light chains and serum N-terminal natriuretic peptide type B decrease simultaneously in association with improvement of survival in AL. Blood. 2006;107:3854–8.
54 Ueno T, Takeda K, Nagata M. Remission of proteinuria and preservation of renal function in patients with renal AA amyloidosis secondary to rheumatoid arthritis. Nephrol Dial Transplant. 2011; doi:10.1093/ndt/gfr357.
55 Nakamura T. Amyloid A amyloidosis secondary to rheumatoid arthritis: pathophysiology and treatment. Clin Exp Rheumatol. 2011;850–6.
56 Lachmann HJ, Quartier P, So A, Hawkins PN. The emerging role of interleukin 1β in autoinflammatory diseases. Arthritis Rheum. 2011;63:314–24.
57 Stankovic Stojanovic K, Delmas Y, Urena Torres P, Peltier J, Pelle G, Jéru I, et al. Dramatic beneficial effect of interleukin-1 inhibitor treatment in patients with familial Mediterranean fever complicated with amyloidosis and renal failure. Nephrol Dial Transplant. 2011 e-pub ahead of print
58 Kofman T, Grimbert P, Canoui-Poitrine F, Zuber J, Garrigue V, Mousson C, et al. Renal transplantation in patients with AA amyloidosis nephropathy: results from a French multicenter study. Am J Transplant. 2011;11:2423–31.
59 Dember LM, Hawkins PN, Hazenberg BP, Gorevic PD, Merlini G, Butrimiene I, et al. Eprodisate for the treatment of renal disease in AA amyloidosis. N Engl J Med. 2007;356:2349–60.
60 Pepys MB, Herbert J, Hutchinson WL, Tennent GA, Lachmann HJ, Gallimore JR, et al. Targeted pharmacological depletion of serum amyloid P component for treatment of human amyloidosis Nature. 2002;417:254–9.
61 Bodin K, Ellmerich S, Kahan MC, Tennent GA, Loesch A, Gilbertson JA, et al. Antibodies to human serum amyloid P component eliminate visceral amyloid deposits. Nature. 2010;468:93–7.
62 Kluve-Beckerman B, Hardwick J, Du L, Benson MD, Monia BP, Watt A, et al. Antisense oligonucleotide suppression of serum amyloid A reduces amyloid deposition in mice with AA amyloidosis. Amyloid. 2011;18:136–46.