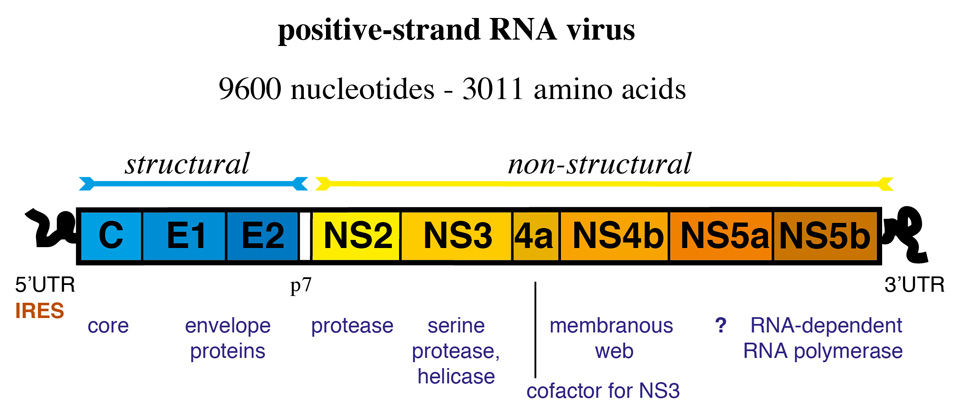
Figure 1
Genetic organisation of hepatitis C virus (HCV). Internal ribosomal entry site (IRES)-mediated translation of the positive-strand RNA genome yields a 3011 long polyprotein that is cleaved into 10 proteins by cellular and viral proteases.
DOI: https://doi.org/10.4414/smw.2012.13586
Chronic hepatitis C (CHC) affects hundreds of millions of people worldwide [1]. Hepatitis C virus (HCV) replicates in hepatocytes. Viral infection provokes an initial immune response that eliminates the virus in 15% to 45% of patients during acute hepatitis C (AHC) [2]. However, in the majority of infected individuals, HCV infection becomes chronic. There is evidence that a strong and multispecific T-cell response is an important host factor for spontaneous viral eradication [3]. More recently, genomewide association studies revealed a very strong association between allelic variants of the interleukin 28B (IL28B) gene locus with spontaneous clearance [4, 5]. This finding provides evidence for a decisive role of the innate immune system in HCV clearance, because the IL28B gene encodes the type III interferon IFNλ3. The induction of type I (IFNα and β) and III (IFNλ) interferons in infected cells is an early and crucial event in the host defence against viruses. How the allelic variants discovered near the IL28B gene can influence the host response to HCV is at present unknown and remains one of the most intriguing questions in the field.
Interferons are not only important for the initial host response to HCV, but remain key components of the immune response during the chronic phase of hepatitis C which lasts decades and can lead to cirrhosis and hepatocellular carcinoma. In a substantial group of patients with CHC the endogenous IFN system in the liver is constantly activated, and hundreds of IFN stimulated genes (ISGs) are strongly expressed in hepatocytes [6–10]. Again, there is an association of genetic variants near the IL28B gene and ISG expression in the liver [11–13], but the molecular pathways that link the IL28B genotype with induction of ISGs are at present unknown. Also, viral escape strategies allowing HCV to persist despite such strong activation of the hepatic IFN system are poorly understood. Most intriguingly, activation of the endogenous IFN system is not only ineffective in clearing HCV, but also strongly inhibits the response to IFN-based therapies [8–10].
Recombinant IFNα was first used in 1986, even before the discovery and cloning of HCV in 1989, for the treatment of chronic non-A/non-B hepatitis, now known as CHC [14]. Since then, IFNα has remained a key component of all therapies against CHC. Initially, unmodified recombinant IFNα2 was injected 3 times a week for 6–12 months. This monotherapy cured 15–25% of patients. In the late 1990s recombinant IFNα2 was combined with ribavirin, improving the sustained virological response rates to 30%-40% [15]. In 2001/2002, pegylated IFNα2 (pegIFNα2) replaced unmodified IFNα2. Because of its longer serum half-life the dosing interval could be increased to one week. Also, it further increased cure rates to 40%–50% [16, 17]. More recently, triple therapies with protease inhibitors (telaprevir or boceprevir), pegIFNα2 and ribavirin were shown to improve response rates in patients infected with HCV genotype 1 to 65%–80% [18–20]. From the early days of CHC treatment up to the present, triple therapies, a patients intrinsic response to injected (peg-)IFNα was the key determinant of treatment failure and success. The search for host and viral factors that determine response to IFNα has so far revealed two major components. First, patients with a constitutive induction of ISGs in the liver during CHC are non-responders [9, 10], and second, IL28B genotype is strongly associated with response to peg IFNα/ribavirin [4, 21–23]. For both components the molecular pathways linking them to non-response are unknown.
HCV was identified as the causative agent of non-A non-B hepatitis in 1989 [24]. HCV is a member of the Flaviviridae family, which includes flaviviruses (yellow fever, dengue and tickborne, encephalitis viruses), the animal pestiviruses (e.g. bovine viral diarrhoea virus) and GB viruses A (GBV-A), GBV-B and GBV-C [25]. Humans are the only natural host and reservoir of HCV. Chimpanzees can be experimentally infected, but natural transmission from one animal to the next has not been described. There are ongoing efforts to develop “humanised” mouse models, but up to now chimpanzees remain the only immunocompetent animal model. HCV replicates in human hepatocytes. The fraction of infected hepatocytes in the liver of patients with CHC is not known, since there are still no reliable and reproducible methods of detecting HCV in liver biopsies. An estimated 1012 virions are released per day from the liver of an infected individual, and same number are cleared daily from the blood [26]. The calculated serum half-life of a virion is 2–3 hours. In a typical infection, serum viral loads range from 104 to 107 IU/mL (1 IU/mL ≈ 2.5 copies/mL).
Figure 1
Genetic organisation of hepatitis C virus (HCV). Internal ribosomal entry site (IRES)-mediated translation of the positive-strand RNA genome yields a 3011 long polyprotein that is cleaved into 10 proteins by cellular and viral proteases.
HCV is a positive, single-stranded RNA virus with a 9.6 kb genome (figure 1). The genome is composed of a 5’-non-coding region (NCR), an open reading frame that encodes 10 viral proteins, and a 3’-NCR. Three structural proteins, the core protein and the two glycoproteins E1 and E2, form the viral particle. The non-structural (NS) proteins include the p7 ion channel, the NS2–3 protease, the NS3 serine protease and RNA helicase, the NS4A polypeptide (a cofactor for NS3 protease), the NS4B protein involved in the formation of the membranous web, the NS5A protein with unknown function, and the NS5B RNA-dependent RNA polymerase [27].
There are 6 HCV genotypes that differ in their nucleotide sequence by 30–35% [28]. Within an HCV genotype subtypes (i.e. HCV genotype 1a, 1b) differ in their nucleotide sequence by 20–25%. Because of the high replicative activity of HCV, and the lack of a proof-reading function of the viral RNA dependent RNA polymerase, there is high genetic variability even within HCV subtypes. The heterogeneous population of HCV genomes that coexist in an infected individual are termed quasispecies. HCV genotypes do not cause different diseases and the natural course of the disease is the same. However, they have different susceptibilities to (peg)IFNα2. Over 75% of patients infected with genotypes 2 and 3 can be cured with pegIFNα2/ribavirin combination therapy, whereas the sustained virological response in genotype 1 infected patients is below 50% [29]. Furthermore, several of the new direct-acting antiviral drugs are effective in specific genotypes only. For example, telaprevir and boceprevir, the two drugs presently approved for CHC treatment in the USA and Europe, are only effective against HCV genotype 1.
Patients with CHC may have nonspecific symptoms including fatigue and malaise, but many patients are asymptomatic. HCV infection is usually detected either through screening of high risk populations (i.e. intravenous drug abuse, history of blood transfusion before 1990), or during a diagnostic workup of elevated liver enzymes. It is estimated that 20–30% of patients with chronic HCV infection will develop cirrhosis over the course of 20–40 years [30–32], but that the majority will never progress to this stage. Cirrhosis is associated with portal hypertension and can lead to ascites, encephalopathy, variceal bleeding, coagulopathy, spontaneous bacterial peritonitis and, most gruesome, to hepatocellular carcinoma (HCC). The yearly incidence of HCC in patients with cirrhosis and active hepatitis C is 2%–5%. HCV associated end-stage cirrhosis is one of the leading motives for liver transplantation in the developed world.
CHC may also be associated with extrahepatic diseases such as mixed cryoglobulinaemia vasculitis, Sicca/Sjögren’s syndrome, thrombocytopenia, insulin resistance and diabetes mellitus, lichen planus, and B-cell lymphoproliferative disorders [33].
The current standard of care for patients infected with HCV genotype 1 consists of triple therapy with an NS3 protease inhibitor (boceprevir or telaprevir), pegIFNα2 and ribavirin. The treatment is given for 6–12 months, depending on previous treatment history, absence or presence of cirrhosis, and the initial response to treatment [34]. All other HCV genotypes are treated with a combination of pegIFNα2 and ribavirin [35]. CHC treatment is undergoing rapid changes. Several dozens of new direct-acting antiviral drugs are presently in clinical development, many of them active against all genotypes [36]. No vaccine against HCV is available. HCV has several strategies for evading the host immune response, including the capacity to undergo antigenic escape and to inhibit the host immune response at several levels. Hence the development of a preventive or therapeutic vaccine remains a major challenge for future HCV research [37, 38].
Interferon was identified more than 50 years ago by Isaacs and Lindenmann during their studies of the phenomenon of viral interference, the ability of an active or inactivated virus to interfere with the growth of an unrelated virus [39]. Today, more than 10 mammalian IFN species and numerous subspecies have been discovered, each with individual properties, but all having antiviral activity [40]. They are currently classified into three groups: type I, type II and type III IFNs. The type I IFNs include all IFNαs, IFNβ, IFNε, IFNκ, IFNω and IFNν [40]. Humans have 12 different IFNαs and a single IFNβ. Type I IFN genes are clustered on the human chromosome 9. Each subtype is encoded by its own gene and regulated by its own promoter, and none of them contain introns [41]. The different IFNαs and IFNβ differ substantially in their specific antiviral activities and in the ratios of antiviral to antiproliferative activities. However, the molecular basis of these differences is not yet known. All type I IFNs bind to the same interferon alpha/beta receptor (IFNAR) which consists of two major subunits: IFNAR1 (the a subunit in the older literature) [42] and IFNAR2c (the βL subunit) [43, 44].
There is only one class II IFN, IFNγ. IFNγ is produced by T lymphocytes when stimulated with antigens or mitogens. IFNγ binds to a distinct receptor, the interferon gamma receptor (IFNGR) consisting of the two subunits IFNGR1 (previously α chain) [45] and IFNGR2 (previously β chain or accessory factor) [46, 47].
The more recently described type III IFNs IFNλ2, IFNλ3 and IFNλ1 are also known as IL28A, IL28B and IL29 respectively [48, 49]. The same as type I IFNs, they are also induced by viral infections [50–52]. They signal through the IFN-λ receptor consisting of the IL-10R2 chain shared with the IL-10 receptor, and a unique IFNλ chain [48, 53].
Cells produce IFNαs, IFNβ and IFNλs in response to infection by a variety of viruses. Unlike bacteria and fungi, which have microbe-specific structures distinguishable from host cell structures, viruses are made predominantly of host-derived components. Given the lack of virus specific proteins or lipids, the cellular receptors that detect viruses have instead evolved to recognise the presence of the viral genome composed of nucleic acids. Two important pathways that detect viral genomes and induce type I and type III IFNs have been discovered and characterised in recent years: the toll-like receptor (TLR) dependent pathway [54, 55] and the cytosolic pathway triggered by binding of viral RNA to the RNA helicases retinoic acid inducible gene-I (RIG-I) and melanoma differentiation antigen 5 (MDA5) [56, 57].
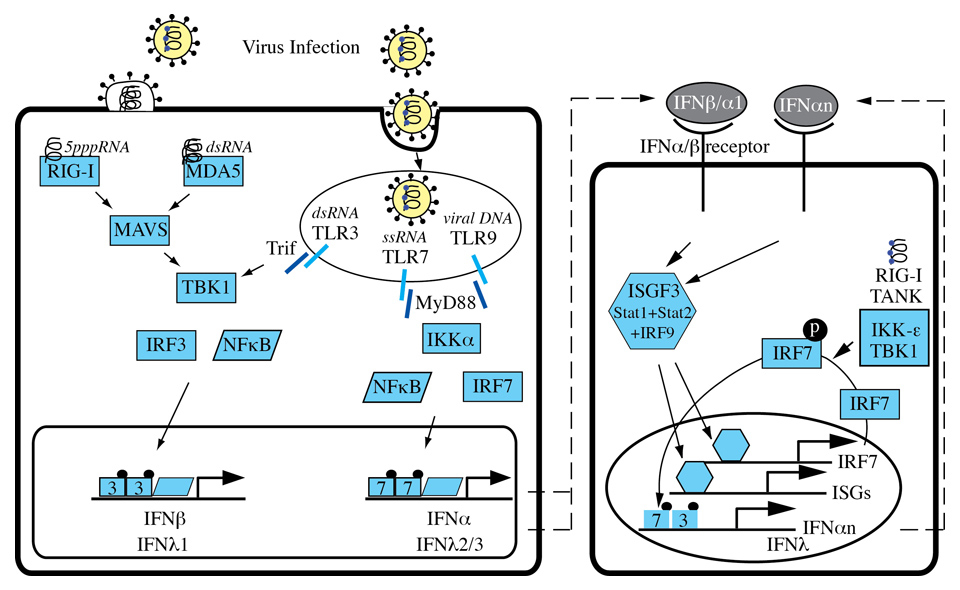
Figure 2
Induction of IFNs.Viral infections are sensed by two important pathways: the toll-like receptor (TLR) dependent pathway and the cytosolic pathway triggered by binding of viral RNA to the RNA helicases retinoic acid inducible gene-I (RIG-I) and melanoma differentiation antigen 5 (MDA5). RIG-I and MDA5 signal through MAVS and TBK1 to activate the transcription factors IRF3 and NFκB. TLR3 signalling depends on the adaptor TRIF to activate TBK, IRF and NFκB, whereas TLR7 and TLR9 use the MyD88-IKKα pathway to activate IFN gene transcription.
TLRs are a family of transmembrane pattern recognition receptors (PRRs) that recognise microbial pathogen associated molecular patterns (PAMPs) and activate the expression of genes involved in inflammatory and immune responses [55]. There are at least 10 human TLRs, and 3 of them are involved in the recognition of viral infections: TLR3, TLR7 and TLR9. TLRs are expressed on various immune cells such as macrophages, dendritic cells, B-cells, but also on fibroblasts and epithelial cells. While TLRs involved in the recognition of bacterial components are expressed on the cell surface, TLR3, TLR7 and TLR9 are localised in intracellular compartments such as endosomes. TLR3 recognises dsRNA (e.g. HCV-RNA) [58], TLR7 detects ssRNA [59, 60] and TLR9 interacts with unmethylated DNA with CpG motifs [61]. TLR activation induces signalling cascades that mainly involve the key transcription factors NF-κB and various interferon regulatory factors (IRFs) (figure 2). Specifically, IRF3 and IRF7 have both distinct and essential roles for virus induced transcriptional activation of IFNβ [62]. IRF3 is constitutively expressed in most cells, whereas IRF7 is expressed at low levels and is strongly expressed only after stimulation of cells with type I IFNs [63]. TLR3 uses the adapter protein Trif and the kinase TBK1 to activate mainly IRF3 in conventional dendritic cells and macrophages, whereas TLR7 and TLR9 induce the expression and secretion of large amounts of type I IFNs in plasmacytoid dendritic cells through the adaptor molecule MyD88 which directly interacts with IRF7 (not IRF3) [64, 65]. The MyD88 pathway requires the IRAK4–IRAK1–IKKα kinase cascade to activate both IRF7 and the NF-κB pathway [66].
The cytosolic pathway of type I and III IFN induction is initiated by recognition of viral 5’triphosphate RNA and dsRNA by RIG-I and MDA5. Binding of viral RNA induces a conformational change of these sensors that results in binding to MAVS (Cardif, VISA, IPS-1), an essential downstream adaptor in the cytosolic pathway [67–70]. Through as yet unidentified mediators, MAVS then propagates the signal to the TBK1 and IKKi kinases that finally activate the transcription factors IRF3 and NF-κB. Activated IRF3 and NF-κB bind to response elements in the promoters of type I and III IFN genes (figure 2).
The initial release of IFNs activates a very powerful positive feedback loop able to produce high local IFN concentrations. IFNs bind to IFN receptors on the same cell or on neighbouring cells, activate the Jak-STAT pathway and further induce IFN gene transcription [63].
The receptor complex for type I IFNs consists of IFNAR1 (the a subunit in the older literature) [42] and IFNAR2c (the βL subunit) [43, 44]. Each receptor subunit constitutively binds to a single specific member of the Janus kinase (Jak) family: IFNAR1 to tyrosine kinase 2 (TYK2) and IFNAR2c to Jak1 (Figure 3). Upon binding of the two chains by type I IFNs, TYK2 and JAK1 transactivate each other by mutual tyrosine phosphorylation, and then initiate a cascade of tyrosine phosphorylation events on the intracellular domains of the receptors and on signal transducer and activator of transcription (STAT) 1, STAT2 and STAT3. The IL-10 receptor binds to Jak1, the IFN-λ chain to Tyk2. IFNGR1 (previously α chain) [45] and IFNGR2 (previously β chain or accessory factor) bind to Jak1 and Jak2, respectively [46, 47].
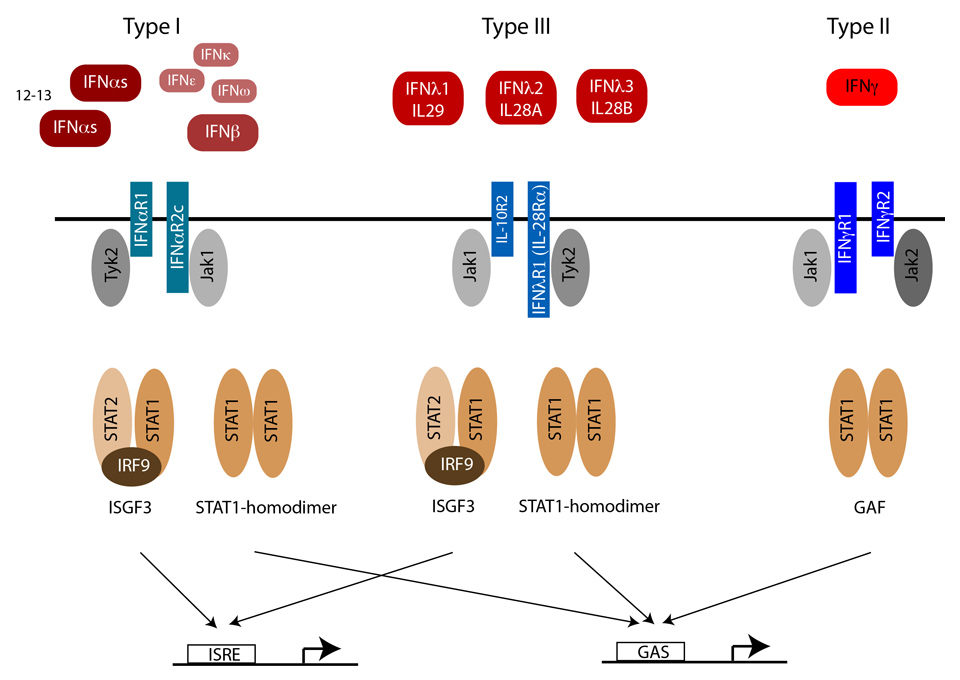
Figure 3
IFN signalling through the Jak-STAT pathway.Type I and III IFNs bind to distinct receptors, but activate the same downstream signalling pathways, and induce a widely overlapping set of genes through the activation of IFN stimulated gene factor 3 (ISGF3) and STAT1 homodimers. IFNγ, the only type II IFN, activates STAT1 homodimers, but not ISGF3, and thereby induces an overlapping but distinct set of ISGs.
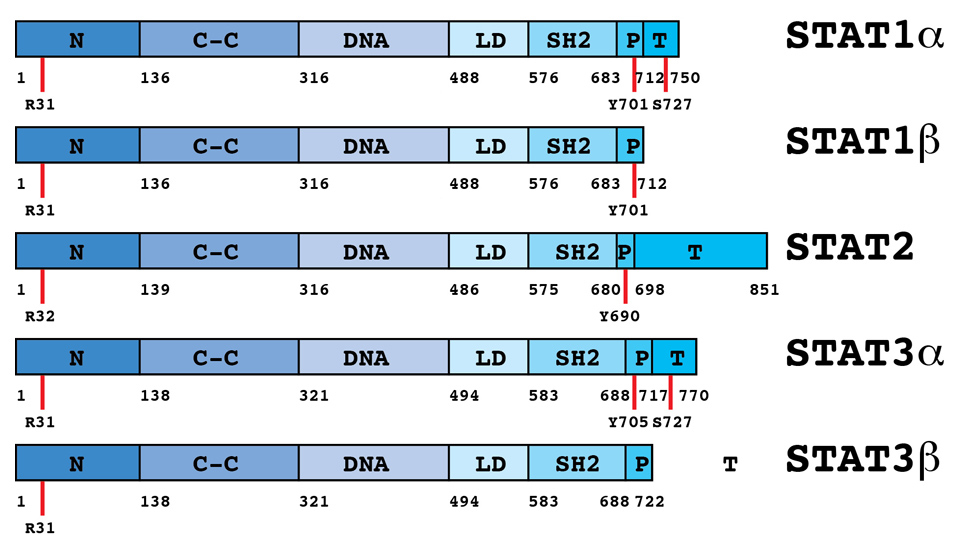
Figure 4
Domain structure of STATs.There are seven STAT genes in the human genome, coding for STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b and STAT6. Differential splicing and posttranslational cleavage can form multiple isoforms. For IFN signaling, STAT1 and STAT2 (and in some cells STAT3) are most important. STATs are between 750 and 850 amino acids long. All STATs share well-defined, structurally and functionally conserved domains including the amino-terminal (N), coiled-coil, DNA-binding, linker, SH2, tyrosine activation, and transcriptional activation domains.
In most cells, type I IFNs activate STAT1, STAT2 and STAT3. STAT1 and STAT2 combine with a third transcription factor, IRF9, to form interferon-stimulated gene factor 3 (ISGF3). ISGF3 binds to interferon stimulated response elements (ISREs) in the promoters of IFN stimulated genes (ISGs). Alternatively, IFN activated STAT1 and STAT3 can form homodimers or STAT1–STAT3 heterodimers. These STAT dimers bind a different class of response elements, the gamma activated sequence (GAS) elements. Once bound to the promoters of ISGs, STATs induce the transcription of genes involved in the generation of an antiviral state [71, 72].
STAT proteins are between 750 and 850 amino acids long. They share well-defined, structurally and functionally conserved domains including the amino-terminal (NH2), coiled-coil, DNA-binding, linker, SH2, tyrosine activation, and transcriptional activation domains (figure 4) [73]. The N-terminal domain is important for homotypic dimerisation of inactive STATs and for cooperative DNA binding to tandem GAS elements [74, 75]. The coiled-coil domain is a protein interaction domain. Binding to GAS elements is provided by the adjacent DNA binding domain. The SH2 domain has a central role for the recruitment of STATs to tyrosine phosphorylated receptors and for dimerisation of activated STATs, and, importantly, provides specificity of signalling through the Jak-STAT pathway [76]. The carboxy-terminal residues constitute the transactivation domain. Alternative splicing at the 3’ end of the gene transcripts generates shorter isoforms of STAT1, 3, 4, 5A and 5B. The shorter isoforms lack a functional transcriptional activation domain, but retain the capacity to occupy specific binding sites in the promoters of target genes. By competing with full length STATs for DNA binding sites, they can inhibit transcriptional activation of target genes, and when overexpressed can be dominant negative regulators of transcription. However, in multimeric complexes with other transcription factors, these short isoforms need not be negative regulators of transcription. STAT1β can combine with STAT2 and IRF9 to form the transcription factor ISGF3, and STAT3β and c-Jun cooperatively bind to an IL-6 responsive promoter element in the α2–macroglobulin gene and activate its transcription [77, 78]. In both cases the transcriptional activation domain is provided by the partner proteins of the short STAT isoforms.
IFN signalling is controlled by a number of negative regulators such as SOCS, USP18, PIAS and TcPTP. Suppressor of cytokine signalling (SOCS) proteins are important negative regulators of Jak-STAT signalling [79]. The family consists of eight members, CIS and SOCS1 to SOCS7. CIS, SOCS1, SOCS2 and SOCS3 are rapidly induced by a large number of cytokines and inhibit cytokine receptors in an early negative feedback loop. Type I IFNs induce SOCS1 and SOCS3 [80], and overexpression experiments have demonstrated that both inhibit IFN signalling through the Jak-STAT pathway [80, 81]. SOCS1-deficient mice develop severe inflammatory disease [82], but are highly resistant to viral infections, most probably due to enhanced type I IFN signalling [83]. SOCS3 simultaneously binds to cytokine receptors and JAK1, JAK2, and TYK2 (but not JAK3), and inhibits the catalytic domain of the kinases [84].
Ubiquitin specific peptidase 18 (USP18/UBP43) is another important negative regulator in type I IFN signalling. USP18/UBP43 was originally identified as a protease cleaving ubiquitin-like modifier ISG15 from target proteins, but was recently found to play a negative regulatory role independently of its ISG-deconjugating ability [85, 86]. UBP43 was reported to inhibit activation of Jak1 by interfering with the binding of Jak1 to IFNAR2c [87]. UBP43 deficient mice show a severe phenotype characterised by brain cell injury, poly-I:C hypersensitivity, and premature death [88, 89]. Interestingly, they are resistant to otherwise fatal cerebral infections with lymphocytic choriomeningitis virus and vesicular stomatitis virus [90].
Protein inhibitor of activated STAT1 (PIAS1) and PIAS3 specifically bind to tyrosine phosphorylated STAT1 and STAT3 respectively, and inhibit DNA binding of STAT dimers [91]. PIAS1 selectively inhibits interferon-inducible genes and is important in innate immunity. As a consequence, PIAS1-deficient mice show increased protection against pathogenic infections [92].
STAT1 is deactivated in the nucleus by dephosphorylation of the tyrosine 701 by T cell protein tyrosine phosphatase (TcPTP) [93]. TcPTP deficient mice develop progressive systemic inflammatory disease, as shown by chronic myocarditis, gastritis, nephritis, sialadenitis and elevated serum IFNγ [94]
It has been known for many years that cultured cells become refractory to IFN within hours and remain unresponsive for up to 3 days [95]. Maximal activation of the IFN signalling pathways is observed within the first two hours of IFN treatment. Continuous exposure to IFN results in “desensitisation” characterised by a return to pretreatment levels of ISG transcription. Moreover, during the 48 to 72 hours following the initial IFNα stimulation of the cells, any further IFN treatment fails to reinduce transcription of ISGs [95].
Refractoriness is observed not only in cultured cells, but also in the liver of mice injected with mouse IFNα [96]. Hepatocytes in vivo become refractory within hours after the first subcutaneous injection of IFNα and remain so for at least 2 days. A systematic analysis of the negative regulators of IFNα signalling revealed that SOCS are responsible for the early inhibition of STAT phosphorylation within the first 2–4 hours, but not for the observed long-term refractoriness. Rather, long-lasting upregulation of USP18/UBP43 was found to be responsible for the observed unresponsiveness of liver cells to prolonged IFNα exposure [96]. Interestingly, IFNβ signalling is not subject to refractoriness in the mouse liver, and, contrary to IFNα, repeated injection of IFNβ elicit strong STAT1 activation after each injection [97]. Likewise, no desensitisation of IFNλ signalling was observed in the intestine of mice (IFNλ signalling could not be investigated in the mouse liver because, unlike human hepatocytes, mouse hepatocytes do not express the IFNλ receptor) [97]. Refractoriness to IFNα could be one of the underlying mechanisms responsible for non-response to pegIFNα/ribavirin therapy observed in patients with CHC who have a constitutively activated endogenous IFN system in the liver.
Interferons exhibit a wide spectrum of biological activities in target cells, including antiviral, immunomodulatory, antiangiogenic and growth-inhibitory effects. They exert their effects mainly through Jak-STAT mediated regulation of gene transcription. However, there are also Jak-STAT independent effects, notably activation of the p38 Map kinase signalling cascade [98, 99], and activation of the phosphatidylinositol 3 kinase – Akt kinase – mTOR/p70 S6 kinase pathway that regulates mRNA translation [100, 101].
Stimulation of cells with type I IFNs usually leads to the induction of several hundred genes (IFN stimulated genes, ISGs), but there are also some genes that are negatively regulated by IFNs [9, 102, 103]. There is considerable variation between different cell types in regard to the number and also the identity of the regulated genes [103]. Gene expression analysis in the human and chimpanzee have shown that systemic administration of (peg) IFNα induces overlapping but clearly distinct sets of genes in liver and peripheral blood mononuclear cells [9, 104]. The mRNA levels of most of the genes are increased 2–10-fold through IFN stimulation, but some genes are induced even more strongly [9]. In the liver, most of the ISGs are upregulated within hours after administration of pegylated IFNα and rapidly downregulated again within the first 8–24 hours [104].
Type I IFN-induced regulation of hundreds of genes establishes an “antiviral state” in the cell [105, 106]. The term “antiviral state” implies protection of the cell against viral infection, but it is a generic term and the lack of precise criteria for its definition reflects the fact that we still have only an elementary understanding of what exactly it is. Indeed, a large number of these regulated genes have as yet unknown functions. Some ISGs have broad antiviral effects. For example, protein kinase R (PKR), a member of the eukaryotic initiation factor 2α (eIF2α) kinase family, phosphorylates eIF2α with a consequent blockade of translation of most cellular and viral mRNAs [107]. Members of the interferon-induced protein with tetratricopeptide repeats (IFIT1 (ISG56) and IFIT2 (ISG54)) also inhibit translation by binding to eIF3 [108]. Another well-studied antiviral effector is 2’-5’ oligoadenylate synthetase (OAS). Both the gene transcription and the enzymatic activity are regulated: the enzymatic activity is stimulated by viral dsRNA, and OAS expression is upregulated several-fold by IFNα. The 2’-5’oligoadenylates produced by activated OAS in turn activate the latent RNA nuclease RNase L, resulting in the degradation of viral and host RNAs [107]. Recently, the ISG15 system has been found to be another broadly active non-specific antiviral effector. ISG15 is one of the most prominent ISGs. It is a ubiquitin-like protein that conjugates to more than 150 cellular target proteins [90, 109–111]. The conjugation is effected by an enzymatic cascade that includes an E1 activating enzyme (UBE1L) [112], an E2 conjugating enzyme (UbcH8) [113, 114], and an E3 ligase (HERC5 and TRIM25) [115, 116]. The conjugation can be reversed by UBP43/USP18 [86]. All these enzymes are induced by type I IFNs. Many of the ISG15 target proteins have important roles in the IFN response, for example Jak1, STAT1, RIG-I, MxA, PKR and RNaseL [110]. Consistent with its role in the IFN system, mice deficient in ISG15 have increased susceptibility to infection with several viruses [117].
In addition to these relatively nonspecific effector systems there are a number of ISGs with activities against distinct classes of virus. For example, the MX proteins have protective effects against influenza and vesicualr stomatitis virus by binding to viral nucleocapsids and the viral polymerase [118], and the members of the APOBEC3 family of cytidine deaminases have activity against HIV [119].
A recent large-scale screen with 380 human ISGs has identified several new antiviral effector molecules. IRF1, C6orf150 (also known as MB21D1), HPSE, RIG-I, MDA5 and IFITM3 were found to be active against different viruses, whereas more targeted antiviral specificity was observed with DDX60, IFI44L, IFI6, IFITM2, MAP3K14, MOV10, NAMPT (also known as PBEF1), OASL, RTP4, TREX1 and UNC84B (also known as SUN2). Mechanistically, most of these antiviral effectors worked through inhibition of translation [120].
Several ISGs have been implicated in the host defense against HCV. Viperin, a member of the radical S-adenosyl methionine domain containing enzymes, inhibits replication in HCV replicon system [121, 122]. PKR and ISG20, a 3’-5’ exonuclease with a strong preference for single-stranded RNA, also strongly inhibit HCV replicons [122].
In addition to their well-known antiviral effects, type I IFNs inhibit cell growth and control apoptosis, activities that affect the suppression of cancer and infection [123]. Different cells in culture exhibit varying degrees of sensitivity to the antiproliferative activity of IFNs. Lymphoblastoid Daudi cells are exquisitely sensitive to the antiproliferative effects of IFNα, which lead to a rapid shutdown of c-myc transcription, possibly through a decrease in the activity of the transcription factor E2F [124]. The antiproliferative effects of IFNα are the rational basis for their use in the treatment of metastatic malignant melanoma and renal cell carcinoma [125].
In order to establish a persistent infection over decades, HCV must evade the host’s immune response, both innate and adaptive. As outlined above, viruses are sensed through TLR-dependent pathways [54, 55] and cytosolic pathways triggered by binding of viral RNA to the RNA helicases RIG-I and MDA5 [56, 57]. HCV has the unique capacity to inactivate essential components of both pathways, i.e. TRIF and MAVS, through the proteolytic activity of its non-structural protein NS3–4A [67, 126]. In vitroand in cell cultures, NS3–4A is very efficient in cleaving TRIF and MAVS. However, the inhibition of IFN-inducing pathways is far from complete in vivo. Transcriptome analysis of liver homogenates from chimpanzees with CHC revealed a strong induction of hundreds of ISGs [127]. The situation is more complex in patients with CHC. 50%-70% of Caucasian patients show very little or no induction of ISGs, whereas the remaining 30%–50% have a permanent high-level expression of hundreds of ISGs [8–10]. A study with paired liver biopsies obtained before and 4 hours after the first injection of pegIFNα in 16 patients undergoing therapy for CHC revealed that patients already with constitutive induction of ISGs before therapy (“pre-activated”) had no significant further increase or qualitative change in ISG expression after pegIFNα, a finding that could explain why all these patients did not respond to the later treatment with pegIFNα /ribavirin [9]. The other group of patients without ISG induction responded to pegIFNα injection with the up-regulation of hundreds of ISGs in the liver, and all of these patients were subsequently cured [9]. The strong association between ISG expression measured in pre-treatment biopsies and non-response to later treatments with pegIFNα /ribavirin has been confirmed in several independent studies [8–10]. By measurement of expression levels of selected ISGs in pre-treatment biopsies it is possible to predict later treatment outcome with high accuracy [11].
An important unresolved question is why activation of the endogenous IFN system in the liver of “pre-activated” patients seems to be ineffective against HCV, whereas induction of the same set of genes in “non pre-activated” patients by injected pegIFNα is highly effective, with cure rates >90% in this group of patients. The difference may arise from the percentage of hepatocytes with ISG induction. In post-treatment biopsies of “non-pre-activated” patients, almost 100% of hepatocytes show nuclear staining for phosphorylated STAT1, whereas the percentage of STAT1 positive nuclei is lower in pre-treatment biopsies of “pre-activated” patients [9]. Assuming that HCV affects 5%–30% of hepatocytes in the liver, the observed ISG induction in liver homogenates may arise from the vast majority of non-infected hepatocytes with intact Jak-STAT signalling. In infected hepatocytes, IFN signaling could be blocked by HCV. There is indeed evidence that HCV, similar to other viruses, interferes with IFN signaling.
One proposed mechanism is the inhibition of STAT1 activation through an upregulation of SOCS3 by HCV core protein, which has been found after transient transfection of HepG2 and Huh7 cells [128, 129]. Another group reported that the expression of HCV proteins in Huh7 cells leads to a proteasome-dependent degradation of STAT1 [130]. A third group, also using HCV protein expression in Huh7 cells, reported normal STAT1 expression and phosphorylation, but an inhibition of nuclear translocation of phosphorylated STAT1 [131]. We have found an inhibition of DNA binding of activated STATs not only in cells transfected with the HCV genome, but also in the liver of HCV transgenic mice, and in liver biopsies of patients with CHC [132, 133]. In all cases, STAT1 protein expression and tyrosine phosphorylation were not impaired. Further investigations of the molecular mechanisms of HCV interference with IFN signalling identified protein phosphatase 2A (PP2A) as an important mediator in the inhibitory pathway [134]. The catalytic subunit of PP2A, PP2Ac, was found to be overexpressed as a result of an endoplasmatic reticulum (ER) stress response induced by HCV protein expression [135]. PP2Ac was overexpressed in cells after HCV protein expression, in liver extracts of HCV transgenic mice, and in liver biopsies of patients with CHC [134]. Further, expression of a constitutively active form of PP2Ac in Huh7 cells resulted in inhibition of STAT1 DNA binding [134]. PP2A can bind directly to protein arginine methyltransferase 1 (PRMT1) and inhibit its enzymatic activity [136]. This inhibition of PRMT1 results in decreased methylation of a number of proteins, including STAT1 [134]. It has been reported that arginine methylation of STAT1 regulates the association of STAT1 with the inhibitor PIAS1 [137], a finding that is still controversial [138]. Nonetheless, we have found that inhibition of PRMT1 by increased expression of PP2Ac leads to increased association of STAT1 with PIAS1, a finding that could well explain the impaired DNA binding of activated STATs in HCV infected cells [134]. Interestingly, treatment of cells with the methyl group donor S-adenosyl-methionine (SAMe) restored normal IFN signalling in cells with HCV protein expression and increased the potency of IFNα in the replicon system [139]. On the basis of these findings we and others conducted clinical studies that showed limited efficacy of SAME given together with pegIFNα and ribavirin in patients with CHC [140, 141].
HCV was able to interfere not only with IFN signalling through the Jak-STAT pathway, but also with the translation of ISG mRNAs to proteins at the ribosomes. Elegant evidence for such a translational block has been obtained in Huh7.5 cells infected with HCV and treated with IFNα. In this system HCV infection did not block the transcriptional induction of ISGs by IFNα [142]. However, HCV infection triggered phosphorylation and activation of the RNA-dependent protein kinase PKR, which inhibits eukaryotic translation initiation factor eIF2 alpha, and thereby cap-dependent translation of cellular mRNAs, but not the IRES-dependent translation of HCV RNA [142].
If and to what extent HCV interference with Jak-STAT signalling, ISG transcription or ISG mRNA translation is occurring in infected hepatocytes in patients with CHC remains to be investigated. One of the main obstacles for answering this important question remains the lack of a reproducible and reliable method to detect HCV RNA or proteins in liver biopsies.
Because of its enormous impact on global health, HCV has been the subject of intense research efforts by academic and industrial research groups. Cloning of the virus in 1989 by Houghton and colleagues allowed in the following decade the expression, purification and high-resolution structure analysis of viral proteins, and the rational design of small inhibitory molecules targeting vital enzymatic activities of these proteins. Several dozens of direct-acting antiviral drugs are in clinical development, and many more in pre-clinical evaluation. There is very reasonable hope for patients with CHC that the therapeutic options will improve significantly in the next decade.
For the vast majority of the estimated over 200 million patients worldwide, these therapies will not be easily obtainable, for economic and logistic reasons. The only hope for an effective control of the HCV epidemic relies on preventive vaccines. Progress in this field has been slow for several reasons. HCV infection is a highly dynamic process driven by a high virus replication rate combined with an error-prone genome replication that produces millions of viral variants each day and facilitates antigenic escape from adaptive immune responses. HCV has also evolved a number of strategies to interfere with signalling pathways and effector systems of the innate and adaptive immune system. A fundamentally improved understanding of these mechanisms will be necessary for the rational design of vaccines. The study of host-virus interactions therefore remains important even in the coming area of highly effective, interferon-free, direct-acting anti-HCV therapies.
1 Lavanchy D. The global burden of hepatitis C. Liver Int. 2009;29(Suppl 1):74–81.
2 Santantonio T, Wiegand J, Gerlach JT. Acute hepatitis C: current status and remaining challenges. J Hepatol. 2008;49(4):625–33.
3 Thimme R, Oldach D, Chang KM, Steiger C, Ray SC, Chisari FV. Determinants of viral clearance and persistence during acute hepatitis C virus infection. J Exp Med. 2001;194(10):1395–406.
4 Rauch A, Kutalik Z, Descombes P, Cai T, Di Iulio J, Mueller T, et al. Genetic variation in IL28B is associated with chronic hepatitis C and treatment failure: a genome-wide association study. Gastroenterology. 2010;138(4):p. 1338–45, 1345 e1-7.
5 Thomas DL, Thio CL, Martin MP, Qi Y, Ge D, O’Huigin C, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature. 2009;461(7265):798–801.
6 MacQuillan GC, Mamotte C, Reed WD, Jeffrey GP, Allan JE. Upregulation of endogenous intrahepatic interferon stimulated genes during chronic hepatitis C virus infection. J Med Virol. 2003;70(2):219–27.
7 Asselah T, Bieche I, Laurendeau I, Paradis V, Vidaud D, Degott C, et al. Liver gene expression signature of mild fibrosis in patients with chronic hepatitis C. Gastroenterology. 2005;129(6):2064–75.
8 Asselah T, Bieche I, Narguet S, Sabbagh A, Laurendeau I, Ripault MP, et al. Liver gene expression signature to predict response to pegylated interferon plus ribavirin combination therapy in patients with chronic hepatitis C. Gut. 2008;57(4):516–24.
9 Sarasin-Filipowicz M, Oakeley EJ, Duong FH, Christen V, Terracciano L, Filipowicz W, et al. Interferon signaling and treatment outcome in chronic hepatitis C. Proc Natl Acad Sci U S A. 2008;105(19):7034–9.
10 Chen L, Borozan I, Feld J, Sun J, Tannis LL, Coltescu C, et al. Hepatic gene expression discriminates responders and nonresponders in treatment of chronic hepatitis C viral infection. Gastroenterology. 2005;128(5):1437–44.
11 Dill MT, Duong FH, Vogt JE, Bibert S, Bochud PY, Terracciano L, et al. Interferon-induced gene expression is a stronger predictor of treatment response than IL28B genotype in patients with hepatitis C. Gastroenterology. 2011;140(3):1021–1031 e10.
12 Honda M, Sakai A, Yamashita T, Nakamoto Y, Mizukoshi E, Sakai Y, et al. Hepatic ISG expression is associated with genetic variation in interleukin 28B and the outcome of IFN therapy for chronic hepatitis C. Gastroenterology. 2010;139(2):499–509.
13 Urban TJ, Thompson AJ, Bradrick SS, Fellay J, Schuppan D, Cronin KD, et al. IL28B genotype is associated with differential expression of intrahepatic interferon-stimulated genes in patients with chronic hepatitis C. Hepatology 2010.
14 Hoofnagle JH, Mullen KD, Jones DB, Rustgi V, Di Bisceglie A, Peters M, et al. Treatment of chronic non-A,non-B hepatitis with recombinant human alpha interferon. A preliminary report. N Engl J Med. 1986;315(25):1575–8.
15 McHutchison JG, Gordon SC, Schiff ER, Shiffman ML, Lee WM, Rustgi VK, et al. Interferon alfa-2b alone or in combination with ribavirin as initial treatment for chronic hepatitis C. Hepatitis Interventional Therapy Group. N Engl J Med. 1998;339(21):1485–92.
16 Manns MP, McHutchison JG, Gordon SC, Rustgi VK, Shiffman M, Reindollar R, et al. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet. 2001;358(9286):958–65.
17 Fried MW, Shiffman ML, Reddy KR, Smith C, Marinos G, Goncales FL, Jr., et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med. 2002;347(13):975–82.
18 Jacobson IM, McHutchison JG, Dusheiko G, Di Bisceglie AM, Reddy KR, Bzowej NH, et al. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med. 2011;364(25):2405–16.
19 Zeuzem S, Andreone P, Pol S, Lawitz E, Diago M, Roberts S, et al. Telaprevir for retreatment of HCV infection. N Engl J Med. 2011;364(25):2417–28.
20 Poordad F, McCone J, Jr., Bacon BR, Bruno S, Manns MP, Sulkowski MS, et al. Boceprevir for untreated chronic HCV genotype 1 infection. N Engl J Med. 2011;364(13):1195–206.
21 Ge D, Fellay J, Thompson AJ, Simon JS, Shianna KV, Urban TJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature. 2009;461(7262):399–401.
22 Suppiah V, Moldovan M, Ahlenstiel G, Berg T, Weltman M, Abate ML, et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nature Genetics. 2009;41(10):1100–4.
23 Tanaka Y, Nishida N, Sugiyama M, Kurosaki M, Matsuura K, Sakamoto N, et al. Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nature Genetics. 2009;41(10):1105–9.
24 Choo QL, Kuo G, Weiner AJ, Overby LR, Bradley DW, Houghton M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science. 1989;244(4902):359–62.
25 Lindenbach BD, Thiel HJ, Rice CM. Flaviviridae: The viruses and their replication, in fields virology. Knipe DM, Howley PM (Editors). Philadelphia: Lippincott-Raven Publishers; 2007. p. 1101–52.
26 Perelson AS, Herrmann E, Micol F, Zeuzem S. New kinetic models for the hepatitis C virus. Hepatology. 2005;42(4):749–54.
27 Moradpour D, Penin F, Rice CM. Replication of hepatitis C virus. Nat Rev Microbiol. 2007;5(6):453–63.
28 Simmonds P, Bukh J, Combet C, Deleage G, Enomoto N, Feinstone S, et al. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology. 2005;42(4):962–73.
29 Hadziyannis SJ, Sette H, Jr., Morgan TR, Balan V, Diago M, Marcellin P, et al. Peginterferon-alpha2a and ribavirin combination therapy in chronic hepatitis C: a randomized study of treatment duration and ribavirin dose. Ann Intern Med. 2004;140(5):346–55.
30 Freeman AJ, Dore GJ, Law MG, Thorpe M, Von Overbeck J, Lloyd AR, et al. Estimating progression to cirrhosis in chronic hepatitis C virus infection. Hepatology. 2001;34(4 Pt 1):809–16.
31 Alter MJ, Kruszon-Moran D, Nainan OV, McQuillan GM, Gao F, Moyer LA, et al. The prevalence of hepatitis C virus infection in the United States, 1988 through 1994. N Engl J Med. 1999;341(8):556–62.
32 Vogt M, Lang T, Frosner G, Klingler C, Sendl AF, Zeller A, et al. Prevalence and clinical outcome of hepatitis C infection in children who underwent cardiac surgery before the implementation of blood-donor screening. N Engl J Med. 1999;341(12):866–70.
33 Jacobson IM, Cacoub P, Dal Maso L, Harrison SA, Younossi ZM. Manifestations of chronic hepatitis C virus infection beyond the liver. Clin Gastroenterol Hepatol. 2010;8(12):1017–29.
34 Treatment of chronic hepatitis C genotype 1 with triple therapy comprising telaprevir or boceprevir. Swiss Med Wkly. 2012;142:w13516.
35 EASL Clinical Practice Guidelines: management of hepatitis C virus infection. J Hepatol. 2011;55(2):245–64.
36 Pockros PJ. Drugs in development for chronic hepatitis C: a promising future. Expert Opin Biol Ther. 2011;11(12):1611–22.
37 Torresi J, Johnson D, Wedemeyer H. Progress in the development of preventive and therapeutic vaccines for hepatitis C virus. J Hepatol. 2011;54(6):1273–85.
38 Houghton M. Prospects for prophylactic and therapeutic vaccines against the hepatitis C viruses. Immunol Rev. 2011;239(1):99–108.
39 Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proceedings of the Royal Society of London. Series B, Containing papers of a Biological character 1957;147(927):258–67.
40 Pestka S. The interferons: 50 years after their discovery, there is much more to learn. J Biol Chem. 2007;282(28):20047–51.
41 Pestka S, Krause CD, Walter MR. Interferons, interferon-like cytokines, and their receptors. Immunol Rev. 2004;202:8–32.
42 Uze G, Lutfalla G, Gresser I. Genetic transfer of a functional human interferon alpha receptor into mouse cells: cloning and expression of its cDNA. Cell. 1990;60(2):225–34.
43 Novick D, Cohen B, Rubinstein M. The human interferon alpha/beta receptor: characterization and molecular cloning. Cell. 1994;77(3):391–400.
44 Lutfalla G, Holland SJ, Cinato E, Monneron D, Reboul J, Rogers NC, et al. Mutant U5A cells are complemented by an interferon-alpha beta receptor subunit generated by alternative processing of a new member of a cytokine receptor gene cluster. Embo J. 1995;14(20):5100–8.
45 Aguet M, Dembic Z, Merlin G. Molecular cloning and expression of the human interferon-gamma receptor. Cell. 1988;55(2):273–80.
46 Soh J, Donnelly RJ, Kotenko S, Mariano TM, Cook JR, Wang N, et al. Identification and sequence of an accessory factor required for activation of the human interferon gamma receptor. Cell. 1994;76(5):793–802.
47 Hemmi S, Bohni R, Stark G, Di Marco F, Aguet M. A novel member of the interferon receptor family complements functionality of the murine interferon gamma receptor in human cells. Cell. 1994;76(5):803–10.
48 Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, Shah NK, et al. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol. 2003;4(1):69–77.
49 Sheppard P, Kindsvogel W, Xu W, Henderson K, Schlutsmeyer S, Whitmore TE, et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat Immunol. 2003;4(1):63–8.
50 Coccia EM, Severa M, Giacomini E, Monneron D, Remoli ME, Julkunen I, et al. Viral infection and Toll-like receptor agonists induce a differential expression of type I and lambda interferons in human plasmacytoid and monocyte-derived dendritic cells. Eur J Immunol. 2004;34(3):796–805.
51 Osterlund PI, Pietila TE, Veckman V, Kotenko SV, Julkunen I. IFN regulatory factor family members differentially regulate the expression of type III IFN (IFN-lambda) genes. J Immunol. 2007;179(6):3434–42.
52 Onoguchi K, Yoneyama M, Takemura A, Akira S, Taniguchi T, Namiki H, et al. Viral infections activate types I and III interferon genes through a common mechanism. J Biol Chem. 2007;282(10):7576–81.
53 Donnelly RP, Sheikh F, Kotenko SV, Dickensheets H. The expanded family of class II cytokines that share the IL-10 receptor-2 (IL-10R2) chain. J Leukoc Biol. 2004;76(2):314–21.
54 Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5(10):987–95.
55 Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124(4):783–801.
56 Yoneyama M, Fujita T. Function of RIG-I-like receptors in antiviral innate immunity. J Biol Chem. 2007;282(21):15315–8.
57 Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5(7):730–7.
58 Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by toll-like receptor 3. Nature. 2001;413(6857):732–8.
59 Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303(5663):1529–31.
60 Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303(5663):1526–9.
61 Bauer S, Kirschning CJ, Hacker H, Redecke V, Hausmann S, Akira S, et al. Human TLR9 confers responsiveness to bacterial DNA via species-specific CpG motif recognition. Proc Natl Acad Sci U S A. 2001;98(16):9237–42.
62 Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, et al. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity. 2000;13(4):539–48.
63 Marie I, Durbin JE, Levy DE. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. Embo J. 1998;17(22):6660–9.
64 Honda K, Yanai H, Mizutani T, Negishi H, Shimada N, Suzuki N, et al. Role of a transductional-transcriptional processor complex involving MyD88 and IRF-7 in Toll-like receptor signaling. Proc Natl Acad Sci U S A. 2004;101(43):15416–21.
65 Kawai T, Sato S, Ishii KJ, Coban C, Hemmi H, Yamamoto M, et al. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat Immunol. 2004;5(10):1061–8.
66 Hoshino K, Sugiyama T, Matsumoto M, Tanaka T, Saito M, Hemmi H, et al. IkappaB kinase-alpha is critical for interferon-alpha production induced by Toll-like receptors 7 and 9. Nature. 2006;440(7086):949–53.
67 Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437(7062):1167–72.
68 Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Molecular Cell. 2005;19(6):727–40.
69 Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122(5):669–82.
70 Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, et al. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol. 2005;6(10):981–8.
71 Darnell JE, Jr. STATs and gene regulation. Science. 1997;277(5332):1630–5.
72 Darnell JE, Jr., Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264(5164):1415–21.
73 Heim MH. The STAT protein family. Signal Transducers and Activators of Transcription (STATs). Activation and Biology. 2003:11–26.
74 Mertens C, Zhong M, Krishnaraj R, Zou W, Chen X, Darnell JE, Jr. Dephosphorylation of phosphotyrosine on STAT1 dimers requires extensive spatial reorientation of the monomers facilitated by the N-terminal domain. Genes & Development. 2006;20(24):3372–81.
75 Vinkemeier U, Moarefi I, Darnell JE, Jr., Kuriyan J. Structure of the amino-terminal protein interaction domain of STAT-4. Science. 1998;279(5353):1048–52.
76 Heim MH, Kerr IM, Stark GR, Darnell JE, Jr. Contribution of STAT SH2 groups to specific interferon signaling by the Jak-STAT pathway. Science. 1995;267(5202):1347–9.
77 Schaefer TS, Sanders LK, Nathans D. Cooperative transcriptional activity of Jun and Stat3 beta, a short form of Stat3. Proc Natl Acad Sci U S A. 1995;92(20):9097–101.
78 Zhang X, Wrzeszczynska MH, Horvath CM, Darnell JE, Jr. Interacting regions in Stat3 and c-Jun that participate in cooperative transcriptional activation. Mol Cell Biol. 1999;19(10):7138–46.
79 Krebs DL, Hilton DJ. SOCS proteins: negative regulators of cytokine signaling. Stem Cells. 2001;19(5):378–87.
80 Song MM, Shuai K. The suppressor of cytokine signaling (SOCS) 1 and SOCS3 but not SOCS2 proteins inhibit interferon-mediated antiviral and antiproliferative activities. J Biol Chem. 1998;273(52):35056–62.
81 Sakamoto H, Kinjyo I, Yoshimura A. The janus kinase inhibitor, Jab/SOCS-1, is an interferon-gamma inducible gene and determines the sensitivity to interferons. Leukemia & lymphoma. 2000;38(1-2):49–58.
82 Alexander WS, Starr R, Fenner JE, Scott CL, Handman E, Sprigg NS, et al. SOCS1 is a critical inhibitor of interferon gamma signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell. 1999;98(5):597–608.
83 Fenner JE, Starr R, Cornish AL, Zhang JG, Metcalf D, Schreiber RD, et al. Suppressor of cytokine signaling 1 regulates the immune response to infection by a unique inhibition of type I interferon activity. Nat Immunol. 2006;7(1):33–9.
84 Babon JJ, Kershaw NJ, Murphy JM, Varghese LN, Laktyushin A, Young SN, et al. Suppression of cytokine signaling by SOCS3: characterization of the mode of inhibition and the basis of its specificity. Immunity 2012.
85 Liu LQ, Ilaria R, Jr., Kingsley PD, Iwama A, van Etten RA, Palis J, et al. A novel ubiquitin-specific protease, UBP43, cloned from leukemia fusion protein AML1-ETO-expressing mice, functions in hematopoietic cell differentiation. Mol Cell Biol. 1999;19(4):3029–38.
86 Malakhov MP, Malakhova OA, Kim KI, Ritchie KJ, Zhang DE. UBP43 (USP18) specifically removes ISG15 from conjugated proteins. J Biol Chem. 2002;277(12):9976–81.
87 Malakhova OA, Kim KI, Luo JK, Zou W, Kumar KG, Fuchs SY, et al. UBP43 is a novel regulator of interferon signaling independent of its ISG15 isopeptidase activity. Embo J. 2006;25(11):2358–67.
88 Ritchie KJ, Malakhov MP, Hetherington CJ, Zhou L, Little MT, Malakhova OA, et al. Dysregulation of protein modification by ISG15 results in brain cell injury. Genes & Development. 2002;16(17):2207–12.
89 Malakhova OA, Yan M, Malakhov MP, Yuan Y, Ritchie KJ, Kim KI, et al. Protein ISGylation modulates the JAK-STAT signaling pathway. Genes & Development. 2003;17(4):455–60.
90 Ritchie KJ, Hahn CS, Kim KI, Yan M, Rosario D, Li L, et al. Role of ISG15 protease UBP43 (USP18) in innate immunity to viral infection. Nat Med. 2004;10(12):1374–8.
91 Liu B, Liao J, Rao X, Kushner SA, Chung CD, Chang DD, et al. Inhibition of Stat1-mediated gene activation by PIAS1. Proc Natl Acad Sci U S A. 1998;95(18):10626–31.
92 Liu B, Mink S, Wong KA, Stein N, Getman C, Dempsey PW, et al. PIAS1 selectively inhibits interferon-inducible genes and is important in innate immunity. Nat Immunol. 2004;5(9):891–8.
93 ten Hoeve J, de Jesus Ibarra-Sanchez M, Fu Y, Zhu W, Tremblay M, David M, et al. Identification of a nuclear Stat1 protein tyrosine phosphatase. Mol Cell Biol. 2002;22(16):5662–8.
94 Heinonen KM, Nestel FP, Newell EW, Charette G, Seemayer TA, Tremblay ML, et al. T-cell protein tyrosine phosphatase deletion results in progressive systemic inflammatory disease. Blood. 2004;103(9):3457–64.
95 Larner AC, Chaudhuri A, Darnell JE, Jr. Transcriptional induction by interferon. New protein(s) determine the extent and length of the induction. J Biol Chem. 1986;261(1):453–9.
96 Sarasin-Filipowicz M, Wang X, Yan M, Duong FH, Poli V, Hilton DJ, et al. Alpha interferon induces long-lasting refractoriness of JAK-STAT signaling in the mouse liver through induction of USP18/UBP43. Mol Cell Biol. 2009;29(17):4841–51.
97 Makowska Z, Duong FH, Trincucci G, Tough DF, Heim MH. Interferon-beta and interferon-lambda signaling is not affected by interferon-induced refractoriness to interferon-alpha in vivo. Hepatology. 2011;53(4):1154–63.
98 Goh KC, Haque SJ, Williams BR. p38 MAP kinase is required for STAT1 serine phosphorylation and transcriptional activation induced by interferons. Embo J. 1999;18(20):5601–8.
99 Li Y, Sassano A, Majchrzak B, Deb DK, Levy DE, Gaestel M, et al. Role of p38alpha map kinase in type I interferon signaling. J Biol Chem. 2004;279(2):970–9.
100 Yang CH, Murti A, Pfeffer SR, Kim JG, Donner DB, Pfeffer LM. Interferon alpha /beta promotes cell survival by activating nuclear factor kappa B through phosphatidylinositol 3-kinase and Akt. J Biol Chem. 2001;276(17):13756–61.
101 Kaur S, Sassano A, Dolniak B, Joshi S, Majchrzak-Kita B, Baker DP, et al. Role of the akt pathway in mRNA translation of interferon-stimulated genes. Proc Natl Acad Sci U S A. 2008;105(12):4808–13.
102 Der SD, Zhou A, Williams BR, Silverman RH. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc Natl Acad Sci U S A. 1998;95(26):15623–8.
103 de Veer MJ, Holko M, Frevel M, Walker E, Der S, Paranjape JM, et al. Functional classification of interferon-stimulated genes identified using microarrays. J Leukoc Biol. 2001;69(6):912–20.
104 Lanford RE, Guerra B, Lee H, Chavez D, Brasky KM, Bigger CB. Genomic response to interferon-alpha in chimpanzees: implications of rapid downregulation for hepatitis C kinetics. Hepatology. 2006;43(5):961–72.
105 van Boxel-Dezaire AH, Rani MR, Stark GR. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity. 2006;25(3):361–72.
106 Stetson DB, Medzhitov R. Type I interferons in host defense. Immunity. 2006;25(3):373–81.
107 Sadler AJ, Williams BR. Interferon-inducible antiviral effectors. Nat Rev Immunol. 2008;8(7):559–68.
108 Taliani G, Gemignani G, Ferrari C, Aceti A, Bartolozzi D, Blanc PL, et al. Pegylated interferon alfa-2b plus ribavirin in the retreatment of interferon-ribavirin nonresponder patients. Gastroenterology. 2006;130(4):1098–106.
109 Narasimhan J, Potter JL, Haas AL. Conjugation of the 15-kDa interferon-induced ubiquitin homolog is distinct from that of ubiquitin. J Biol Chem. 1996;271(1):324–30.
110 Zhao C, Denison C, Huibregtse JM, Gygi S, Krug RM. Human ISG15 conjugation targets both IFN-induced and constitutively expressed proteins functioning in diverse cellular pathways. Proc Natl Acad Sci U S A. 2005;102(29):10200–5.
111 Giannakopoulos NV, Luo JK, Papov V, Zou W, Lenschow DJ, Jacobs BS, et al. Proteomic identification of proteins conjugated to ISG15 in mouse and human cells. Biochem Biophys Res Commun. 2005;336(2):496–506.
112 Yuan W, Krug RM. Influenza B virus NS1 protein inhibits conjugation of the interferon (IFN)-induced ubiquitin-like ISG15 protein. Embo J. 2001;20(3):362–71.
113 Kim KI, Giannakopoulos NV, Virgin HW, Zhang DE. Interferon-inducible ubiquitin E2, Ubc8, is a conjugating enzyme for protein ISGylation. Mol Cell Biol. 2004;24(21):9592–600.
114 Zhao C, Beaudenon SL, Kelley ML, Waddell MB, Yuan W, Schulman BA, et al. The UbcH8 ubiquitin E2 enzyme is also the E2 enzyme for ISG15, an IFN-alpha/beta-induced ubiquitin-like protein. Proc Natl Acad Sci U S A. 2004;101(20):7578–82.
115 Zou W, Zhang DE. The interferon-inducible ubiquitin-protein isopeptide ligase (E3) EFP also functions as an ISG15 E3 ligase. J Biol Chem. 2006;281(7):3989–94.
116 Wong JJ, Pung YF, Sze NS, Chin KC. HERC5 is an IFN-induced HECT-type E3 protein ligase that mediates type I IFN-induced ISGylation of protein targets. Proc Natl Acad Sci U S A. 2006;103(28):10735–40.
117 Lenschow DJ, Lai C, Frias-Staheli N, Giannakopoulos NV, Lutz A, Wolff T, et al. IFN-stimulated gene 15 functions as a critical antiviral molecule against influenza, herpes, and Sindbis viruses. Proc Natl Acad Sci U S A. 2007;104(4):1371–6.
118 Arnheiter H, Skuntz S, Noteborn M, Chang S, Meier E. Transgenic mice with intracellular immunity to influenza virus. Cell. 1990;62(1):51–61.
119 Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002;418(6898):646–50.
120 Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature. 2011;472(7344):481–5.
121 Helbig KJ, Lau DT, Semendric L, Harley HA, Beard MR. Analysis of ISG expression in chronic hepatitis C identifies viperin as a potential antiviral effector. Hepatology. 2005;42(3):702–10.
122 Jiang D, Guo H, Xu C, Chang J, Gu B, Wang L, et al. Identification of three interferon-inducible cellular enzymes that inhibit the replication of hepatitis C virus. J Virol. 2008;82(4):1665–78.
123 Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. How cells respond to interferons. Annu Rev Biochem. 1998;67:227–64.
124 Melamed D, Tiefenbrun N, Yarden A, Kimchi A. Interferons and interleukin-6 suppress the DNA-binding activity of E2F in growth-sensitive hematopoietic cells. Mol Cell Biol. 1993;13(9):5255–65.
125 Friedman RM. Clinical uses of interferons. Br J Clin Pharmacol. 2008;65(2):158–62.
126 Li K, Foy E, Ferreon JC, Nakamura M, Ferreon AC, Ikeda M, et al. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc Natl Acad Sci U S A. 2005;102(8):2992–7.
127 Bigger CB, Guerra B, Brasky KM, Hubbard G, Beard MR, Luxon BA, et al. Intrahepatic gene expression during chronic hepatitis C virus infection in chimpanzees. J Virol. 2004;78(24):13779–92.
128 Bode JG, Ludwig S, Ehrhardt C, Albrecht U, Erhardt A, Schaper F, et al. IFN-alpha antagonistic activity of HCV core protein involves induction of suppressor of cytokine signaling-3. Faseb J. 2003;17(3):488–90.
129 Kawaguchi T, Yoshida T, Harada M, Hisamoto T, Nagao Y, Ide T, et al. Hepatitis C virus down-regulates insulin receptor substrates 1 and 2 through up-regulation of suppressor of cytokine signaling 3. Am J Pathol. 2004;165(5):1499–508.
130 Lin W, Choe WH, Hiasa Y, Kamegaya Y, Blackard JT, Schmidt EV, et al. Hepatitis C virus expression suppresses interferon signaling by degrading STAT1. Gastroenterology. 2005;128(4):1034–41.
131 Melen K, Fagerlund R, Nyqvist M, Keskinen P, Julkunen I. Expression of hepatitis C virus core protein inhibits interferon-induced nuclear import of STATs. J Med Virol. 2004;73(4):536–47.
132 Heim MH, Moradpour D, Blum HE. Expression of hepatitis C virus proteins inhibits signal transduction through the Jak-STAT pathway. J Virol. 1999;73(10):8469–75.
133 Blindenbacher A, Duong FH, Hunziker L, Stutvoet ST, Wang X, Terracciano L, et al. Expression of hepatitis c virus proteins inhibits interferon alpha signaling in the liver of transgenic mice. Gastroenterology. 2003;124(5):p. 1465–75.
134 Duong FH, Filipowicz M, Tripodi M, La Monica N, Heim MH. Hepatitis C virus inhibits interferon signaling through up-regulation of protein phosphatase 2A. Gastroenterology. 2004;126(1):263–77.
135 Christen V, Treves S, Duong FH, Heim MH. Activation of endoplasmic reticulum stress response by hepatitis viruses up-regulates protein phosphatase 2A. Hepatology. 2007;46(2):558–65.
136 Duong FH, Christen V, Berke JM, Penna SH, Moradpour D, Heim MH. Upregulation of protein phosphatase 2Ac by hepatitis C virus modulates NS3 helicase activity through inhibition of protein arginine methyltransferase 1. J Virol. 2005;79(24):15342–50.
137 Mowen KA, Tang J, Zhu W, Schurter BT, Shuai K, Herschman HR, et al. Arginine methylation of STAT1 modulates IFNalpha/beta-induced transcription. Cell. 2001;104(5):731–41.
138 Meissner T, Krause E, Lodige I, Vinkemeier U. Arginine methylation of STAT1: a reassessment. Cell. 2004;119(5):587–9; discussion 589–90.
139 Duong FH, Christen V, Filipowicz M, Heim MH. S-adenosylmethionine and betaine correct hepatitis C virus induced inhibition of interferon signaling in vitro. Hepatology. 2006;43(4):796–806.
140 Feld JJ, Modi AA, El-Diwany R, Rotman Y, Thomas E, Ahlenstiel G, et al. S-adenosyl methionine improves early viral responses and interferon-stimulated gene induction in hepatitis C nonresponders. Gastroenterology 2010.
141 Filipowicz M, Bernsmeier C, Terracciano L, Duong FH, Heim MH. S-adenosyl-methionine and betaine improve early virological response in chronic hepatitis C patients with previous nonresponse. PLoS ONE. 2010;5(11):e15492.
142 Garaigorta U, Chisari FV. Hepatitis C virus blocks interferon effector function by inducing protein kinase R phosphorylation. Cell Host Microbe. 2009;6(6):513–22.
Funding / potential competing interests: No financial support and no other potential conflict of interest relevant to this article was reported.