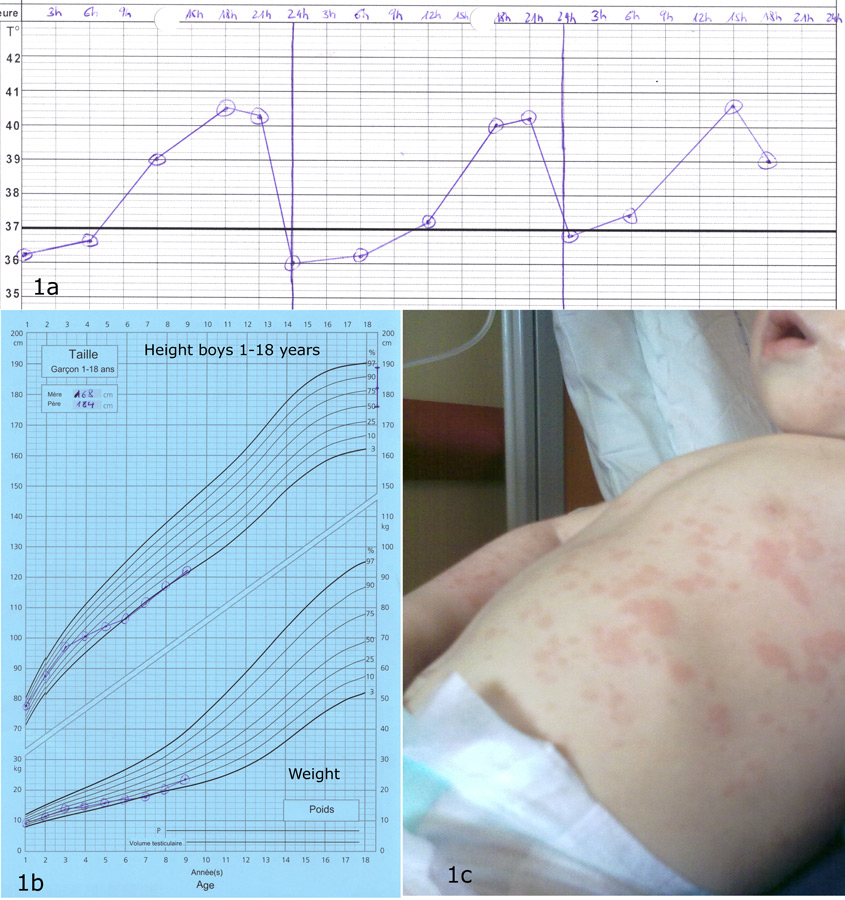
Figure 1
(1a) Typical temperature curve with one fever peak per day; (1b) short stature in a boy with SoJIA, beginning of disease at 2.5 years; (1c) rash in SoJIA.
DOI: https://doi.org/10.4414/smw.2012.13582
In 1897, while working as a registrar at the Hospital for Sick Children at Great Ormond Street in London, Sir George Frederick Still described a series of patients with three patterns of childhood arthritis, one of which came to be known later as Still’s disease, corresponding to what we today call SoJIA [1]. Some authors still refer to this disease entity as Still’s disease, although the international terminology of JIA has been accepted worldwide. Of note, Diamand-Berger had already described the disease in his medical thesis in 1891, six years before Sir G.F. Still. Since chronic arthritis in children and adolescents is clearly a heterogeneous condition, it became evident that classification criteria were mandatory to define homogeneous categories of patients for clinical research. Since 1995, a group of experts under the patronage of the International League against Rheumatism proposed a classification based on an international consensus. Seven subtypes of JIA were defined, and this replaced the previous categories of juvenile chronic arthritis (JCA) and juvenile rheumatoid arthritis (JRA) [2].
JIA is one of the most common paediatric chronic illnesses: the prevalence rate varies widely in the different series (between 10 and 400 cases/100,000), and the yearly incidence varies between 2 and 19.5 cases for 100,000 children [3]. SoJIA as a subtype includes about 10–15% of all JIA patients, but the percentage is higher when severe JIA cases are considered [4, 5]. At onset, it is clinically well distinguished from other forms of JIA by the prominence of extra-articular features such as spiking fevers, a salmon-coloured skin rash, lymphadenopathy and serositis [6]. However, diagnosis is often challenging, since the disease can mimic infections and malignancies. Unlike the other forms of JIA, the affected child looks very ill, especially during fever spikes, and may present with a life-threatening complication known as macrophage activation syndrome (MAS).
SoJIA patients are classically treated first with non-steroidal anti-inflammatory drugs, and if the inflammation is not controlled, high dose corticosteroids are used. Before the use of biotherapies, the disease was difficult to control in a significant number of patients and long lasting steroid treatment induced severe side effects, mainly growth failure and osteoporosis. The clinical course at later stages of SoJIA is highly variable, with about a third of cases having a monocyclic course, a third having a relapsing-remitting course, and another third having continuous disease activity. Systemic features tend to subside over time, while progressive and destructive arthritis can develop [5].
A 30-month-old healthy girl presented with a 3 week history of daily spiking fevers, fatigue, weight loss and poor general conditions. She refused to walk and wanted to be carried by her parents. The presence of hand and feet swelling led to an admission to the emergency room. On examination, she was febrile at 38.6 °C, with an ill appearance, and irritable. General examination was normal except for a discrete erythematous rash on the trunk and the extremities, and enlarged cervical lymph nodes. On musculoskeletal examination, almost all joints from the cervical spine to the toes were involved, with swelling, tenderness, pain and reduced mobility. The patient’s laboratory tests showed major systemic inflammation with anaemia, neutrophilia, thrombocytosis and hyperferritinaemia. Liver enzymes were normal; total IgG levels were increased, and autoantibodies were all negative. An echocardiography showed a moderate pericardial effusion and could exclude coronary involvement. Infection and neoplasia were excluded by laboratory tests and radiological examinations. Based on the clinical presentation and after exclusion of other causes, we could confirm the diagnosis of SoJIA. The patient was treated with high doses of steroids (intravenous followed by oral) and indomethacin. She showed a transient clinical improvement (absence of fever and rash), but persistence of polyarthritis and laboratory inflammation. Methotrexate subcutaneously was added without any effect on joint inflammation. Tocilizumab (anti-IL-6 receptor antibody) was therefore introduced with infusions every 2 weeks. During the following weeks, both systemic and joint inflammation improved significantly and the steroid dose could be decreased. The patient could walk and play again, but many joints remained inflamed, in particular the knees and wrists which were injected with corticosteroids. One year after diagnosis, the patient was treated with tocilizumab, methotrexate and prednisone (0.5 mg/kg/d), laboratory tests were within the normal range, but 27 joints still showed signs of inflammation. We then tried to change the treatment from tocilizumab to etanercept for a better control of joint inflammation. This did not allow a better control of joint inflammation, and the inflammatory markers rose again leading to a new change of the biotherapy to tocilizumab again. We present here a case with a particularly severe course of SoJIA, which significantly improved with a newly recognised treatment for this disease. As illustrated by this case, the course of illness had a major impact on the quality of life for the patient and her family. In complement to the medical care, the family had access to a hotline for counselling and support from our nursing team, who also provided help for the injections. This support was useful to achieve a good therapeutic compliance and provide the necessary service from the network.
The diagnosis of SoJIA should be considered in children or adolescents with typical spiking fevers associated with arthritis or polyarthralgia. This is often a challenging diagnosis since many other causes for the fever need to be excluded. SoJIA may present at any age and there is no clear gender predominance. A seasonal variation for disease onset was suspected, but not confirmed in large studies. The typical picture of SoJIA presents with a very ill child, who is febrile, anaemic and in pain [5–7]. Figure 1 illustrates some of the most important features and complications of the disease.
Figure 1
(1a) Typical temperature curve with one fever peak per day; (1b) short stature in a boy with SoJIA, beginning of disease at 2.5 years; (1c) rash in SoJIA.
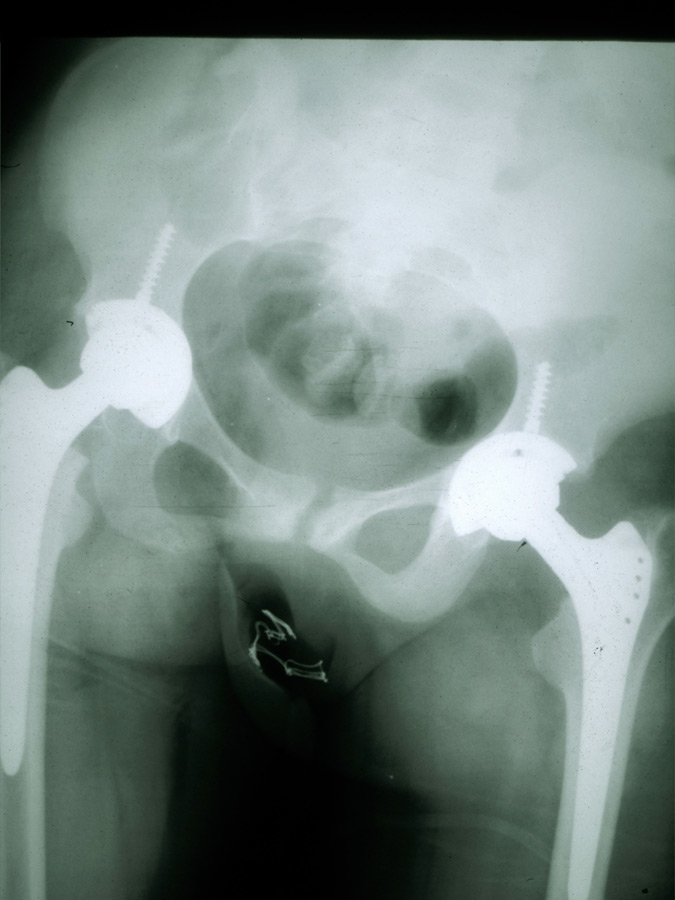
Figure 2
Hip replacement in a patient with SoJIA.
Any joint may be affected at disease onset, and involvement may be oligo- or polyarticular, but arthritis tends to increase in severity over time. In about a third of the patients, only arthralgia are reported at the onset of disease, and the presence of clear arthritis may be observed even years after the occurrence of systemic symptoms [4]. The typical spiking fever goes up to 39° or 40° once or twice daily and returns rapidly to 37° or below [6]. A persistent fever over 24 hours should suggest other diagnoses, or MAS complicating SoJIA [8]. An erythematous macular rash (salmon-pink coloured) often accompanies the fever; it is usually localised over the trunk and the proximal extremities, tends to be migratory, and may be urticarial or sometimes pruritic [9, 10]. Enlargement of the lymph nodes, the liver and/or the spleen may occur. In less than 10% of the patients a pericarditis will develop, often asymptomatic and only rarely inducing a tamponade [4, 11]. The two major complications of SoJIA are MAS and secondary amyloidosis [12]. MAS is a severe, potentially life-threatening complication characterised by the excessive activation of well-differentiated macrophages, resulting in persistent fever with generalised malaise, hepatosplenomegaly, lymphadenopathy, liver failure, intravascular coagulation, and neurological involvement. Laboratory tests show a decrease of the inflammatory parameters and a relative hypofibrinogenemia (often normal, rather than elevated levels due to systemic inflammation), elevated liver enzymes, hemocytopenia, high ferritin levels and increased serum triglycerides [13, 14]. This complication, resembling reactive hemophagocytic lymphohistiocytosis, can be life-threatening and needs to be quickly recognised and treated to avoid a fatal outcome. The diagnosis of MAS associated with active SoJIA may be difficult: specific diagnostic guidelines have been developed to help the physician [13]. Secondary amyloidosis is observed in patients with treatment-resistant disease evolving over many years and can lead to renal insufficiency due to parenchymal deposition of serum amyloid A [15]. This rare complication should not be seen anymore in the future due to a better control of systemic inflammation with the new biological therapies. Other complications include a destructive polyarthritis (fig. 2) and growth retardation which is related to the disease activity and, in some patients, a prolonged corticosteroid therapy.
The diagnosis of SoJIA is based on internationally recognised classification criteria (table 1) [2]. Since the presence of arthritis is mandatory, the diagnosis can only be suspected at disease onset in patients without arthritis. JIA is defined by the presence of arthritis for at least 6 weeks with an onset before 16 years of age, after exclusion of all other causes. According to these classification criteria, four different conditions, listed in table 1, will exclude SoJIA. These exclusion criteria are controversial in particular those considering the family history of the patient; indeed, a patient may develop SoJIA even if a close relative is affected by a different disease such as psoriasis or spondylarthritis. Although SoJIA is considered as part of JIA, it is a disease distinct from the other forms: different clinical presentation, pathophysiology and response to treatment. The oligoarticular and polyarticular forms of JIA are considered as autoimmune diseases and often present with autoantibodies (antinuclear antibodies or rheumatoid factor). However, SoJIA is closer to the group of autoinflammatory diseases and the detection of positive autoantibodies is uncommon [16–18]. Adult-onset Still’s disease is an entity similar to SoJIA occurring in adults. Different sets of criteria are used for diagnosis and the most commonly used are the criteria of Yamaguchi [19]. The presence of clear arthritis is not mandatory in contrast to the childhood classification, allowing the inclusion of all cases affected by the disease. We believe that the criteria for SoJIA should be adapted in order to integrate the recent findings suggesting the affiliation to the group of autoinflammatory diseases and to standardise the classification for both the paediatric and the adult form of this condition.
The main diagnostic challenge for SoJIA is the absence of a pathognomonic clinical feature or laboratory marker. In consequence, the clinician has to consider a wide differential diagnosis (see table 2) and perform a large number of investigations to exclude such alternative possibilities. Laboratory and radiological work-up will depend on disease presentation: an atypical clinical picture and in particular the lack of clear arthritis will lead to broader investigations. Many different infections will be considered, such as occult bacterial infection, brucellosis, Lyme disease, cat scratch disease, tuberculosis, infectious mononucleosis. Malignancies, for example leukaemia, lymphoma and neuroblastoma, may mimic the symptoms of SoJIA, and need to be excluded, if they are clinically suspected, with a bone marrow aspirate. Fever and arthritis may also suggest other inflammatory diseases, like systemic lupus erythematosus, systemic vasculitis, Kawasaki disease, Behçet disease and inflammatory bowel disease. Acute rheumatic fever (ARF) is a more common differential diagnosis in developing countries and will follow a streptococcal A infection; the pattern of arthritis is however acute and migratory and the episode is self-limiting. Recurrent fever syndromes can sometimes present with osteoarticular involvement associated with fever, but the pattern of fever, the clinical picture and the family history will help to distinguish them from SoJIA.
Laboratory investigations will first evaluate the level of inflammation. Levels of erythrocyte sedimentation rate and C-reactive protein are usually very high. A complete blood count is mandatory to exclude lymphoproliferative diseases; it will typically show an inflammatory anaemia, a leukocytosis with neutrophil predominance and a thrombocytosis. A protein produced by neutrophils, myeloid reactive proteins (MRP), is present at very high serum levels in SoJIA, much higher than in other inflammatory illnesses [20]. In the future, this new laboratory test may help to distinguish SoJIA from other febrile conditions. Ferritin is often increased, but very high serum levels will suggest MAS. The immunological status is typically characterised by the absence of autoantibodies.
| Table 1: Classification criteria for SoJIA. |
| Arthritis with or preceding daily fever with a duration of at least 2 weeks accompanied by at least 1 of the following symptoms during the first 6 months. |
| – Erythematous rash; – Lymphadenopathy; – Hepatomegaly and/or splenomegaly; – Serositis. |
| Exclusion criteria |
| – Psoriasis or history of psoriasis in the patient or first degree relative; – Arthritis in a boy (6 year-old or more) positive for HLA-B27; – Ankylosing spondylitis, enthesitis with arthritis, sacro-ileitis and inflammatory bowel disease, Reiter syndrome or acute anterior uveitis in a first degree relative; – Presence of rheumatoid factor type IgM confirmed in an interval of at least 3 months apart. |
| Table 2: Differential diagnosis of SoJIA. |
| Patients <5 years |
| – Bacterial infections; – Malaria; – Leukemia; – Neuroblastoma; – Viral infections; – Kawasaki syndrome; – Hereditary recurrent fever syndromes or other auto-inflammatory syndromes with beginning in early childhood; – Blau syndrome; – Sweet syndrome; – PFAPA (periodic fever, adenopathy, pharyngitis, aphtosis). |
| Patient >5 years |
| – All the above mentioned; – Takayasu syndrome; – Connective tissue diseases; – Rheumatic fever; – Familial Mediterranean Fever or other periodic fever syndromes (TRAPS); – Behçet disease; – Polyarteritis nodosa; – Castleman syndrome; – Inflammatory bowel disease. |
It is now becoming increasingly clear that the markedly distinct clinical presentation of the systemic form of JIA is associated with unique immunologic abnormalities. Several lines of evidence suggest that the role of the adaptive immune system in SoJIA may be rather limited compared to the other JIA types, while the contribution of the innate immunity may be much more prominent [16, 21]. The development of classic autoimmunity is typically associated with the presence of autoreactive antigen-specific T lymphocytes (due to failure of self-tolerance mechanisms), high-titer autoantibodies, and strong MHC class II associations. On the contrary, auto-inflammatory disorders (for example Familial Mediterranean Fever) show abnormalities in innate immunity pathways, lack MHC associations and autoantibodies or antigen specific T cells. Predominance of monocytes and neutrophils rather than lymphocytes as effector cells is another important feature of these diseases. Abnormalities in the innate immunity play a major role in the pathogenesis of SoJIA, which should be viewed as an auto-inflammatory rather than a classic autoimmune disease. Indeed, many features of SoJIA are similar to those seen in auto-inflammatory syndromes, for example presence of fever, multisystem involvement, absence of autoantibodies, and a lack of MHC Class II associations (unlike other forms of JIA). However, a subset of patients with SoJIA may develop autoimmune features in the long-term, such as ANCA-associated glomerulonephritis [22].
The cytokines critical to the perpetuation of the inflammatory process in the systemic form of JIA also appear to be different from those in other JIA categories. Clinical and translational studies suggest the pivotal role for two potent pro-inflammatory cytokines: Interleukin-1 (IL-1) and Interleukin -6 (IL-6) [18]. Indeed, in SoJIA, aberrant activation of phagocytes leads to the secretion of pro-inflammatory cytokines such as IL-1, IL-6 and IL-18, in addition to other inflammatory proteins such as S100A8, S100A9 and S100A12 [17, 20, 23–27].
Although the source of excess IL-1 in this disorder is still obscure, elegant studies from Virginia Pascual’s group in Dallas have shown a major role of this cytokine [18], and response to IL-1 inhibition has been demonstrated in several studies including a recent multicenter French trial (whereas anti-TNF therapy offers only limited benefit in SoJIA patients) [28–31]. Of note, compelling evidence for the involvement of IL-1 in this disorder comes from the efficacy of IL-1 blockage with Anakinra in clinical trials.
The group from Dallas has developed two complementary approaches using microarray technology: (a) analysis of transcriptional patterns in patients’ peripheral blood mononuclear cells; and (b) analysis of the transcriptional changes induced by adding serum from active patients to healthy peripheral blood mononuclear cells (PBMCs). Serum from SoJIA patients induced transcription of various innate immunity genes, including IL-1β, in PBMCs obtained from healthy individuals. Moreover, the authors analysed leukocyte gene expression profiles of paediatric SoJIA patients and compared them to healthy controls and to subjects with other febrile illnesses. Genes differentially expressed in SoJIA patients compared with healthy children were identified, including 12 genes which accurately classified an independent set of SoJIA patients with systemic disease. Transcripts that changed significantly in patients undergoing IL-1 blockade were also identified. Thus, according to these studies, signatures can be used to distinguish SoJIA from other febrile illnesses and to assess response to anti-cytokine therapy [32].
The aforementioned French study [31] was a double blind, placebo-controlled trial conducted on patients with severe, persistently active disease. Its aim was to evaluate the efficacy and safety of Anakinra, as well as to assess treatment effect on surrogate markers of SoJIA activity and gene expression profile. The primary objective was to compare the efficacy of a one-month treatment with Anakinra (2 mg/kg subcutaneously daily, maximum 100 mg) to a placebo between 2 groups of 12 patients each. Response was defined by a 30% improvement of paediatric ACR core-set criteria for JIA, in addition to resolution of fever and systemic symptoms, and a decrease of at least 50% of both C-reactive protein and erythrocyte sedimentation rate. Secondary objectives included tolerance and efficacy assessment over 12 months, pharmacokinetic analyses, treatment effect on blood gene expression, anti-pneumococcal response, serum amyloid A and ferritin levels, and percentage of glycosylated ferritin. At one month, there was a significant difference in the response rate between patients treated with Anakinra and placebo. The number of adverse events, mainly pain related to injections, was similar between both groups. Ten patients from the placebo group switched to Anakinra at Month 1 and nine were responders at Month 2. Blood gene expression profiling analyses showed a SoJIA signature at inclusion that rapidly wore off upon initiation of Anakinra treatment, allowing to distinguish patients treated with Anakinra from patients treated with placebo. Despite being a relatively small study, this represents the first placebo-controlled trial of Anakinra in SoJIA; results should be confirmed in larger scale studies. Of interest, an up-regulation of type I IFN-regulated transcripts, which is not a feature of untreated SoJIA, was induced in the majority of Anakinra-treated patients. The molecular and cellular basis for an IL-1 β/type I IFN cross-regulation in SoJIA remains unknown, although it is known that type I IFN decreases IL-1β and induces IL-1Ra production by PBMCs in vitro. A similar cross-regulation has been described for type I IFN and TNF (patients with rheumatoid arthritis and Crohn’s disease treated with TNF antagonists develop anti-nuclear Abs and some of them display symptoms of systemic lupus erythematosus, a type I IFN-mediated disease). Whether the upregulation of type I IFN-inducible genes in Anakinra-treated patients could have clinical consequences through the triggering of adaptive immunity remains to be determined.
However, results have not been uniformly positive; it seems that there are two different populations with regard to IL-1 response [33], and moreover, the seemingly reduced success of IL-1 inhibition in subjects with long-standing disease suggests that later in the pathological process other mechanisms might be involved.
The role of another important cytokine, Interleukin-6 (IL-6), had been proposed by De Benedetti and colleagues in the past [24, 25]. IL-6 is markedly elevated in both peripheral blood and synovial fluid of patients with SoJIA, and the levels of IL-6 expression appear to correlate with the overall clinical activity of the disease and with distinctive clinical features such as thrombocytosis, microcytic anaemia, growth retardation and osteopenia. Furthermore, studies of the unique quotidian fever pattern of systemic JIA show that IL-6 concentrations rise and fall in concert with the temperature spikes [34]. Tocilizumab is a humanised anti-human interleukin-6 (IL-6) receptor monoclonal antibody of the IgG1 subclass produced by recombinant DNA technology that inhibits the function of IL-6. Clinical studies with tocilizumab have been successful in SoJIA, with one placebo-controlled Japanese trial already published [35] and an international phase III study already completed.
In these cases therapeutic experience has taught us a lot about disease pathogenesis and the importance of these two cytokines has been confirmed by proving the clinical efficacy of blocking their activity. This effect does not seem to be as important in other types of JIA.
IL-18, originally described as an IFN-γ inducing factor mainly produced by activated macrophage lineage cells, stimulates a variety of inflammatory responses. Serum levels of IL-18 have been found to be elevated in patients with SoJIA [26, 36–38] even during inactive disease phases when concentrations of other cytokines normalised [39]. Moreover, it has been shown that the mechanism of the impaired NK cell function that is present in SoJIA involves a defect in the IL-18 receptor beta-phosphorylation [40].
With regard to the calcium-binding proteins of the S100 family, they are known to be secreted specifically by activated phagocytes including monocytes, macrophages and neutrophils. These proteins (S100A8, A9, and A12) are also known as myeloid-related proteins (MRPs). In SoJIA, high circulating levels of these molecules are found, much more than in other inflammatory conditions including different forms of arthritis; hopefully they might also be useful as biomarkers for prognosis [20].
The recent insights into the pathophysiological mechanisms in SoJIA have led to major changes in the management of this disease. Until recently, the treatment algorithms in SoJIA included mainly corticosteroids and methotrexate. Since their introduction, TNF-inhibitors have also been frequently used in these patients [41–43]. Thalidomide [44] and even autologous stem cell transplantation have been used in patients with particularly severe disease. However, methotrexate and anti-TNF [45] seem to be less effective for SoJIA than for other JIA categories. With advancing knowledge on disease pathogenesis, two cytokines have been the target of modern biologic treatment of SoJIA, and clinical experience has now shown us that biologics blocking IL-1 and IL-6 are very effective.
The most relevant experiences with the use of anakinra have already been summarised in the previous paragraph; other reports have followed and confirmed its efficacy. A recent multicenter paper has also suggested that, if given as a first-line drug, anakinra leads to rapid resolution of systemic symptoms and arthritis [46]; however the retrospective nature of this study and the lack of a control group make it difficult to draw firm conclusions. Canakinumab, a fully human anti-interleukin-1β (anti-IL-1β) monoclonal antibody, is another new IL-1 inhibiting agent currently studied in SoJIA in the USA and Europe. Results of a phase II study have just been published [47], while those of a phase III trial should be available soon. Rilonacept (or IL-1 Trap) is a fusion protein consisting of the two human IL1 receptor extracellular domains and the Fc portion of human IgG1. It incorporates the extracellular domains of both receptor chains required for IL-1 signalling within a single molecule: the IL-1 type I receptor and the IL-1 receptor accessory protein. Due to this, the IL-1 Trap molecule might be a more efficient inhibitor of in vivo IL-1 signalling than anakinra. Recently, Rilonacept has been proven to be effective in familial cold auto-inflammatory syndromes [48], and a phase III trial in SoJIA is in progress.
The anti-IL-6 receptor antibody (Tocilizumab) has been studied in several trials, and the drug is now approved in the U.S. and Europe for the treatment of active SoJIA [35, 49].
The optimal treatment for resistant cases has to be tailored to individual patients; in case of failure of NSAIDs and a short course of corticosteroids ± methotrexate (or in case of corticodependance) referral to a specialised paediatric rheumatology centre is recommended. Although the American College of Rheumatology has released treatment recommendations [50], and a consensus conference by members of the Childhood Arthritis & Rheumatology Research Alliance has also been published recently [51], head-to-head trials are not available and therefore comparing the efficacy of different biologics is difficult. Hence, the decision of which biologic therapy to administer in a specific situation cannot be made by using standardised protocols.
Table 3 summarises the different treatment used for SoJIA.
| Table 3: Main treatments used for Systemic-onset JIA. | |
| Treatment | Standard dose |
| NSAIDS | |
| Indomethacin | 2–3 mg/kg/d in 2–3 divided doses |
| Naproxen | 20 mg/kg/d in 2 divided doses |
| Ibuprofen | 30–40 mg/kg/d in 3–4 divided doses |
| Diclofenac | 3 mg/kg/d in 2–3 divided doses |
| Acetylsalicylic acid | 75–100 mg/kg/d in 4 divided doses |
| Second-line drugs | |
| Methotrexate | 10–15 mg/m2 once weekly po or subq |
| Etanercept | 0.4 mg/kg twice/weekly subq |
| Thalidomide | 3–5 mg/kg/d po |
| Anakinra | 2 mg/kg/d subq |
| Canakinumab | 4 mg/kg subq/monthly |
| Tocilizumab | 8 mg/kg/every 2 weeks i.v. |
SoJIA is a severe disorder, most likely belonging to the group of auto-inflammatory diseases. Recently, it has been shown that cytokines play a major role in its pathogenesis, and that blocking these molecules may allow a good control of systemic and joint inflammation. These drugs have transformed the prognosis of SoJIA patients by preventing the damage due to the inflammatory process and to side effects, in particular of the chronic use of high doses of corticosteroids. SoJIA remains a disease with a long course, often over many years, and the risk of complications due to the illness or the medications is still significant. Consequently, patients with SoJIA need to be cared for by a specialised medical team, used to this condition and to biotherapies. Additionally, the impact on the quality of life of the child and the family is major, and will require the support of a multidisciplinary team, including nurses and physical therapists. The recent progresses in the understanding of SoJIA have changed the life of this orphan disease and for our patients. However, SoJIA still represents a significant burden and will require our future efforts in research and clinical care to further improve the daily life and the outcome of our young patients.
1 Still GF. On a form of chronic joint disease in children. Med Chir Trans. 1897;80:47–60 49.
2 Petty RE, Southwood TR, Manners P, Baum J, Glass DN, Goldenberg J, et al. International league of associations for rheumatology classification of juvenile idiopathic arthritis: Second revision, edmonton, 2001. J Rheumatol. 2004;31:390–2.
3 Woo P, Wedderburn LR. Juvenile chronic arthritis. Lancet. 1998;351:969–73.
4 Schneider R, Laxer RM. Systemic onset juvenile rheumatoid arthritis. Baillieres Clin Rheumatol. 1998;12:245–71.
5 Woo P. Systemic juvenile idiopathic arthritis: Diagnosis, management, and outcome. Nat Clin Pract Rheumatol. 2006;2:28–34.
6 Behrens EM, Beukelman T, Gallo L, Spangler J, Rosenkranz M, Arkachaisri T, et al. Evaluation of the presentation of systemic onset juvenile rheumatoid arthritis: Data from the Pennsylvania systemic onset juvenile arthritis registry (Pasojar). J Rheumatol. 2008;35:343–8.
7 Feldman BM, Birdi N, Boone JE, Dent PB, Duffy CM, Ellsworth JE, et al. Seasonal onset of systemic-onset juvenile rheumatoid arthritis. J Pediatr. 1996;129:513–8.
8 Behrens EM, Beukelman T, Paessler M, Cron RQ. Occult macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis. J Rheumatol. 2007;34:1133–8.
9 Calabro JJ, Marchesano JM. Rash associated with juvenile rheumatoid arthritis. J Pediatr. 1968;72:611–9.
10 Schaller J, Wedgwood RJ. Pruritus associated with the rash of juvenile rheumatoid arthritis. Pediatrics. 1970;45:296–8.
11 Goldenberg J, Ferraz MB, Pessoa AP, Fonseca AS, Carvalho AC, Hilario MO, Atra E. Symptomatic cardiac involvement in juvenile rheumatoid arthritis. Int J Cardiol. 1992;34:57–62.
12 Hadchouel M, Prieur AM, Griscelli C. Acute hemorrhagic, hepatic, and neurologic manifestations in juvenile rheumatoid arthritis: Possible relationship to drugs or infection. J Pediatr. 1985;106:561–6.
13 Davi S, Consolaro A, Guseinova D, Pistorio A, Ruperto N, Martini A, et al. An international consensus survey of diagnostic criteria for macrophage activation syndrome in systemic juvenile idiopathic arthritis. J Rheumatol. 2011;38:764–8.
14 Grom AA, Passo M. Macrophage activation syndrome in systemic juvenile rheumatoid arthritis. J Pediatr. 1996;129:630–2.
15 David J. Amyloidosis in juvenile chronic arthritis. Clin Exp Rheumatol. 1991;9:73–8.
16 Ramanan AV, Grom AA. Does systemic-onset juvenile idiopathic arthritis belong under juvenile idiopathic arthritis? Rheumatology. (Oxford) 2005;44:1350–3.
17 Frosch M, Roth J. New insights in systemic juvenile idiopathic arthritis – from pathophysiology to treatment. Rheumatology. (Oxford) 2008;47:121–5.
18 Pascual V, Allantaz F, Arce E, Punaro M, Banchereau J. Role of interleukin-1 (il-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J Exp Med. 2005;201:1479–86.
19 Yamaguchi M, Ohta A, Tsunematsu T, Kasukawa R, Mizushima Y, Kashiwagi H, et al. Preliminary criteria for classification of adult Still's disease. J Rheumatol. 1992;19:424–30.
20 Wittkowski H, Frosch M, Wulffraat N, Goldbach-Mansky R, Kallinich T, Kuemmerle-Deschner J, et al. S100a12 is a novel molecular marker differentiating systemic-onset juvenile idiopathic arthritis from other causes of fever of unknown origin. Arthritis Rheum. 2008;58:3924–31.
21 Vastert SJ, Kuis W, Grom AA. Systemic JIA: New developments in the understanding of the pathophysiology and therapy. Best Pract Res Clin Rheumatol. 2009;23:655–64.
22 Belot A, Bader-Meunier B, Niaudet P, Salomon R, Prieur AM, Noel LH, et al. ANCA-Associated Glomerulonephritis in Systemic-Onset Juvenile Idiopathic Arthritis. Am J Kidney Dis. 2012;59:439–43.
23 Frosch M, Ahlmann M, Vogl T, Wittkowski H, Wulffraat N, Foell D, et al. The myeloid-related proteins 8 and 14 complex, a novel ligand of toll-like receptor 4, and interleukin-1beta form a positive feedback mechanism in systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2009;60:883–91.
24 De Benedetti F, Martini A. Is systemic juvenile rheumatoid arthritis an interleukin 6 mediated disease? J Rheumatol. 1998;25:203–7.
25 de Benedetti F, Massa M, Robbioni P, Ravelli A, Burgio GR, Martini A. Correlation of serum interleukin-6 levels with joint involvement and thrombocytosis in systemic juvenile rheumatoid arthritis. Arthritis Rheum. 1991;34:1158–63.
26 Maeno N, Takei S, Imanaka H, Yamamoto K, Kuriwaki K, Kawano Y, et al. Increased interleukin-18 expression in bone marrow of a patient with systemic juvenile idiopathic arthritis and unrecognized macrophage-activation syndrome. Arthritis Rheum. 2004;50:1935–8.
27 Fujii T, Nojima T, Yasuoka H, Satoh S, Nakamura K, Kuwana M, et al. Cytokine and immunogenetic profiles in japanese patients with adult still’s disease. Association with chronic articular disease. Rheumatology. (Oxford) 2001;40:1398–404.
28 Lequerre T, Quartier P, Rosellini D, Alaoui F, De Bandt M, Mejjad O, et al. Interleukin-1 receptor antagonist (anakinra) treatment in patients with systemic-onset juvenile idiopathic arthritis or adult onset Still disease: Preliminary experience in France. Ann Rheum Dis. 2008;67:302–8.
29 Woo P. Anakinra treatment for systemic juvenile idiopathic arthritis and adult onset Still disease. Ann Rheum Dis. 2008;67:281–2.
30 Verbsky JW, White AJ. Effective use of the recombinant interleukin 1 receptor antagonist anakinra in therapy resistant systemic onset juvenile rheumatoid arthritis. J Rheumatol. 2004;31:2071–5.
31 Quartier P, Allantaz F, Cimaz R, Pillet P, Messiaen C, Bardin C, et al. A multicentre, randomised, double-blind, placebo-controlled trial with the interleukin-1 receptor antagonist anakinra in patients with systemic-onset juvenile idiopathic arthritis (ANAJIS trial). Ann Rheum Dis. 2011;70:747–54.
32 Allantaz F, Chaussabel D, Stichweh D, Bennett L, Allman W, Mejias A, et al. Blood leukocyte microarrays to diagnose systemic onset juvenile idiopathic arthritis and follow the response to IL-1 blockade. J Exp Med. 2007;204:2131–44.
33 Gattorno M, Piccini A, Lasiglie D, Tassi S, Brisca G, Carta S, et al. The pattern of response to anti-interleukin-1 treatment distinguishes two subsets of patients with systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2008;58:1505–15.
34 Prieur AM, Roux-Lombard P, Dayer JM. Dynamics of fever and the cytokine network in systemic juvenile arthritis. Rev Rhum Engl Ed. 1996;63:163–70.
35 Yokota S, Imagawa T, Mori M, Miyamae T, Aihara Y, Takei S, et al. Efficacy and safety of tocilizumab in patients with systemic-onset juvenile idiopathic arthritis: A randomised, double-blind, placebo-controlled, withdrawal phase III trial. Lancet. 2008;371:998–1006.
36 Lotito AP, Campa A, Silva CA, Kiss MH, Mello SB. Interleukin 18 as a marker of disease activity and severity in patients with juvenile idiopathic arthritis. J Rheumatol. 2007;34:823–30.
37 Lin YT, Wang CT, Gershwin ME, Chiang BL. The pathogenesis of oligoarticular/polyarticular vs systemic juvenile idiopathic arthritis. Autoimmun Rev. 2011;10:482–9.
38 Jelusic M, Lukic IK, Tambic-Bukovac L, Dubravcic K, Malcic I, Rudan I, et al. Interleukin-18 as a mediator of systemic juvenile idiopathic arthritis. Clin Rheumatol. 2007;26:1332–4.
39 Shimizu M, Yokoyama T, Yamada K, Kaneda H, Wada H, Wada T, et al. Distinct cytokine profiles of systemic-onset juvenile idiopathic arthritis-associated macrophage activation syndrome with particular emphasis on the role of interleukin-18 in its pathogenesis. Rheumatology. (Oxford) 2010;49:1645–53.
40 de Jager W, Vastert SJ, Beekman JM, Wulffraat NM, Kuis W, Coffer PJ, et al. Defective phosphorylation of interleukin-18 receptor beta causes impaired natural killer cell function in systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2009;60:2782–93.
41 Russo RA, Katsicas MM. Clinical remission in patients with systemic juvenile idiopathic arthritis treated with anti-tumor necrosis factor agents. J Rheumatol. 2009;36:1078–82.
42 Quartier P, Taupin P, Bourdeaut F, Lemelle I, Pillet P, Bost M, et al. Efficacy of etanercept for the treatment of juvenile idiopathic arthritis according to the onset type. Arthritis Rheum. 2003;48:1093–101.
43 Kimura Y, Pinho P, Walco G, Higgins G, Hummell D, Szer I, et al. Etanercept treatment in patients with refractory systemic onset juvenile rheumatoid arthritis. J Rheumatol. 2005;32:935–42.
44 Lehman TJ, Schechter SJ, Sundel RP, Oliveira SK, Huttenlocher A, Onel KB. Thalidomide for severe systemic onset juvenile rheumatoid arthritis: A multicenter study. J Pediatr. 2004;145:856–7.
45 Otten MH, Prince FH, Armbrust W, ten Cate R, Hoppenreijs EP, Twilt M, et al. Factors associated with treatment response to etanercept in juvenile idiopathic arthritis. JAMA. 2011;306:2340–7.
46 Nigrovic PA, Mannion M, Prince FH, Zeft A, Rabinovich CE, van Rossum MA, et al. Anakinra as first-line disease modifying therapy in systemic juvenile idiopathic arthritis. Arthritis Rheum. 2011;63:545–55.
47 Ruperto N, Quartier P, Wulffraat N, Woo P, Ravelli A, Mouy R, et al. A phase II study to evaluate dosing and preliminary safety and efficacy of canakinumab in systemic juvenile idiopathic arthritis with active systemic features. Arthritis Rheum. 2012;64:557–67.
48 Gillespie J, Mathews R, McDermott MF. Rilonacept in the management of cryopyrin-associated periodic syndromes (CAPS). J Inflamm Res. 2010;3:1–8.
49 Woo P, Wilkinson N, Prieur AM, Southwood T, Leone V, Livermore P, et al. Open label phase II trial of single, ascending doses of MRA in caucasian children with severe systemic juvenile idiopathic arthritis: Proof of principle of the efficacy of IL-6 receptor blockade in this type of arthritis and demonstration of prolonged clinical improvement. Arthritis Res Ther. 2005;7:R1281–1288.
50 Beukelman T, Patkar NM, Saag KG, Tolleson-Rinehart S, Cron RQ, DeWitt EM, et al. 2011 American College of Rheumatology recommendations for the treatment of juvenile idiopathic arthritis: Initiation and safety monitoring of therapeutic agents for the treatment of arthritis and systemic features. Arthritis Care Res. (Hoboken) 2011;63:465-82.
51 Dewitt EM, Kimura Y, Beukelman T, Nigrovic PA, Onel K, Prahalad S, et al. Consensus treatment plans for new-onset systemic juvenile idiopathic arthritis. Arthritis Care Res. 2012 Jan 30 [Epub ahead of print].
Funding / potential competing interests: No financial support and no other potential conflict of interest relevant to this article were reported.