Air pollution and epigenetics: effects on SP-A and innate host defence in the lung
DOI: https://doi.org/10.4414/smw.2012.13579
Patricia
Silveyra, Joanna
Floros
Summary
An appropriate immune and inflammatory response is key to defend against harmful agents present in the environment, such as pathogens, allergens and inhaled pollutants, including ozone and particulate matter. Air pollution is a serious public health concern worldwide, and cumulative evidence has revealed that air pollutants contribute to epigenetic variation in several genes, and this in turn can contribute to disease susceptibility. Several groups of experts have recently reviewed findings on epigenetics and air pollution [1–6]. Surfactant proteins play a central role in pulmonary host defence by mediating pathogen clearance, modulating allergic responses and facilitating the resolution of lung inflammation. Recent evidence indicates that surfactant proteins are subject to epigenetic regulation under hypoxia and other conditions. Oxidative stress caused by ozone, and exposure to particulate matter have been shown to affect the expression of surfactant protein A (SP-A), an important lung host defence molecule, as well as alter its functions. In this review, we discuss recent findings in the fields of epigenetics and air pollution effects on innate immunity, with the focus on SP-A, and the human SP-A variants in particular. Their function may be differentially affected by pollutants and specifically by ozone-induced oxidative stress, and this in turn may differentially affect susceptibility to lung disease.
Introduction
Over the last several decades, it has become apparent that air pollution is a serious public health concern, ubiquitous throughout the world. Several studies have identified a strong link between exposure to ambient air pollution and increased morbidity and mortality, mainly due to adverse health outcomes, the result of pollutant-induced inflammation and oxidative stress, as well as cardiopulmonary failure [7–10]. Special attention has been put into the role of epigenetics in mediating not only genetic and physiological responses to certain environmental insults, but also in regulating underlying susceptibility to environmental stressors [11, 12].
Air pollution is defined as contamination of the atmosphere by gaseous, liquid, and/or particulate waste (or its by-products) that can cause harm or discomfort to humans or other living organisms, and/or cause damage to the environment. Gaseous contaminants include mainly oxides of nitrogen, carbon and sulphur, as well as volatile organic compounds and ozone. Particulate matter refers to a complex mixture of solid and liquid small particles containing acids (nitrates, sulphates, etc.), organic chemicals, metals, and/or soil or dust particles that can cause serious cardiovascular damage once inhaled [13]. Disease susceptibility is influenced by a number of genetic and non-genetic factors, most of which may have a different level of impact at different stages of life. Epigenetic mechanisms control gene expression without affecting the DNA sequence itself, via mechanisms that range from structural changes in chromatin structure (e.g., DNA methylation, histone modifications) to post-transcriptional gene regulation (e.g., chromatin-mediated regulation of alternative splicing, microRNA-mediated repression of gene expression) [14–16]. Epigenetic mechanisms can change genome function under exogenous influence, and allow the stable propagation of gene activity states among cell generations, although this is not the case for all genes subjected to epigenetic changes.
A number of studies have shown that air pollutants can cause epigenetic changes that may result in altered pulmonary function, increased risk of respiratory infection and lung disease [17, 18]. Innate immunity molecules and cells in contact with the external environment through the inhaled air, and which are also the first line of defence in response to inhaled pollutants, pathogens and various irritants, are likely to have their function and regulation affected by air pollutants. Correlations among genetic variations and mutations of the surfactant protein (SP) genes with disease susceptibility or pathogenesis have been reported [19–23], with some evidence for the lung host defence molecules SP-A and SP-D, that epigenetic changes may differentially alter their expression and/or function [24]. Of interest, air pollutants such as ozone and particulate matter can influence SP-A expression and function, therefore affecting disease susceptibility to lung infection and other health conditions [25–27]. Whether these pollutant-induced changes involve epigenetic regulation remains to be determined.
Air pollution and health outcomes
A number of contaminants from several natural and non-natural sources are present in ambient air. These include various biological agents, as well as particulate matter of varied size and composition, inorganic and organic volatile compounds such as ozone, carbon monoxide, nitrogen dioxide and sulphur dioxide. These are referred to as “criteria pollutants” by the Environmental Protection Agency (EPA), and have been shown to impact health outcomes, including neuropsychological development in children, as well as to increase the risk of death from all causes, affecting both morbidity and mortality rates [28–31]. Pollutants have also been shown to increase the risk of cardiovascular and respiratory illnesses and pulmonary inflammation, and to affect the susceptibility to respiratory infections, asthma, lung cancer and chronic obstructive pulmonary disease (COPD) [32–34]. Moreover, air particulates, ozone, and diesel exhaust particles are known to form reactive oxygen species, such as superoxide anion, hydrogen peroxide and hydroxyl radicals, which can impair protein function, compromise immune function, exacerbate airway inflammation and hyper-reactivity, and alter pulmonary function, resulting in an increased incidence of lung disease and mortality [8, 17, 35–38]. Ozone, for example, has been shown to affect pulmonary innate immunity associated with impaired host defence [35, 39, 40], increase susceptibility to infection [41, 42], and alter levels of inflammatory mediators [43] and macrophage function [41, 42, 44, 45]. Moreover, several studies have demonstrated that many functions of the innate immune molecule SP-A are also affected by exposure to ozone [26, 43, 46, 47].
Host defence, epigenetics and pollution
Pollutants can affect the lung by altering its immune response and airway inflammation. Susceptibility to air pollutants differs among individuals, as exemplified by several diseases and conditions (e.g., asthma) in which both genetic and non-genetic factors seem to play a role in the individual response to ambient air pollution [48, 49]. Epigenetics refers to changes in the genome that are not coded by the genomic sequence itself, but ultimately affect the expression of gene transcripts, and determine potentially heritable changes in gene expression. Epigenetic modifications involve changes in the DNA, by methylation of cytosine residues located in dinucleotide CpG sites commonly within gene promoters, as well as modifications in chromatin structural proteins. Acetylation and methylation of histones results in an altered ability of the transcriptional machinery to interact with a particular DNA sequence. While acetylation of histones on specific lysine residues is generally associated with gene activation, methylation of histones can either repress or activate mRNA synthesis. More recently, additional levels of epigenetic control have been described at the post-transcriptional regulation of gene expression. These include a) miRNA-mediated repression of translation, by direct interaction of small non-coding single-stranded RNA molecules (miRNAs) with the mRNA 3’UTR, and b) regulation of mRNA splicing by interaction of histones with specific regions of the pre-mRNA, particularly at intron-exon junctions [16].
It was originally proposed that once established, epigenetic modifications are maintained and inherited throughout many generations, but recent findings have determined that this is not the case for all genes and organisms. However, experimental evidence in animal, plant and fungal models has demonstrated trans-generational inheritance of epigenetic marks [50, 51]. In general, DNA methyltransferases are responsible for maintaining the methylation pattern from parental to daughter DNA strands upon cell division, and most cells have their epigenetic marks fixed when they differentiate or exit the cell cycle. However, in certain situations such as disease, or in normal development, these epigenetic marks are removed and re-established in a process noted as “reprogramming” [52]. Of all the epigenetic modifications mentioned above, DNA methylation holds a higher potential of being transmitted through generations, despite the reprogramming events mentioned. It has been demonstrated that transcriptional repression triggered by DNA methylation is also linked to chromatin modification, and the addition of methyl groups to DNA is coupled to histone methylation and deacetylation [53]. Thus, histone modifications can also be viewed as (indirectly) heritable. It must be noted that epigenetic regulation may also indirectly affect the expression of genes, by affecting the genes that encode transcription factors and control their abundance and availability to interact with regulatory sequences.
Several reviews have discussed the relationships between epigenetics and the environment [1–6]. Some of the mechanisms known to trigger epigenetic changes include diet, aging, chronic inflammation, stress, infections caused by diverse agents, hormones and endocrine disruptors, as well as the effects of the environment discussed above [54]. Both aging and diet have been shown to alter the general pattern of methylation in CpG islands, and thus affect disease susceptibility. For example, hypermethylation of tumour suppressor genes and some hormone receptors has been correlated with cancer risk [55]. On the other hand, global DNA hypomethylation has been found to occur with the development of cancer, some autoimmune diseases, and other age-related diseases [54]. The specific effects of epigenetic changes in innate immunity molecules are discussed in detail in the following paragraphs.
Epigenetics and innate immunity
Epigenetic changes affecting gene expression patterns that ultimately affect the overall immune response have already been described [56]. These include changes in genes that express pattern recognition receptors, signalling molecules, cytokines and other molecules involved in both innate and adaptive immunity processes [15, 57].
Innate immunity refers to the initial response of nonspecific defence mechanisms triggered upon exposure to an antigen. As the first line of defence, innate immunity components are continuously subjected to and affected by environmental insults. Epigenetic changes have been described in a number of innate immunity related molecules, and correlations with disease susceptibility and health outcomes have been established. For example, dendritic cells, which are essential components of innate immunity and in the initiation of adaptive immunity, express pattern recognition receptors that aim to detect pathogen-associated molecular patterns. These include toll-like receptors and c-type lectin receptors that have been shown to interact with SP-A and modulate inflammatory responses [58]. As dendritic cells are mainly localised in tissues exposed to environmental insults, they can be subject to epigenetic changes induced in response to various pollutants. Such changes, via their effects on gene expression, can affect susceptibility to infection as well as other pathological challenges.
Surfactant protein A and innate immunity
Lung alveolar type II cells synthesise and secrete pulmonary surfactant, a lipoprotein complex essential for life, whose main function is to reduce surface tension in the alveoli and prevent the lungs from collapsing. Some of the surfactant proteins are important modulators of the innate immune response, and inflammatory processes [59–61]. In particular, SP-A, the most abundant protein of surfactant, has been shown to enhance phagocytosis and chemotaxis of alveolar macrophages, modulate the generation of reactive oxygen species, induce proliferation of immune cells, and stimulate pro-inflammatory cytokine production [62–65].
Human SP-A is encoded by two functional genes, SFTPA1 (SP-A1) and SFTPA2 (SP-A2). Genetic variants of SP-A1 and SP-A2, consisting of single nucleotide polymorphisms (SNPs), haplotypes, and other variations have been associated with acute and chronic lung disease throughout life in several populations and study groups [19, 23, 66]. Furthermore, several diseases and complications are correlated with altered SP-A protein levels and functionality. Patients with diseases such as CF and pneumonia commonly have reduced levels of SP-A [23, 67–70]. However very little, if any, is known about epigenetic changes in SP-A genes or variants that may occur preceding or during the course of such diseases. Numerous in vitro and in vivo studies have shown functional, structural, biophysical and biochemical differences among the two gene products and their variants, including phagocytosis of bacteria by alveolar macrophages in the presence or absence of environmental pollutants such as ozone [26, 59, 71].
The role of SP-A in innate immunity has been extensively studied. SP-A has the ability to bind and agglutinate a wide range of pathogens and allergens, including bacteria, fungi, viruses and other non-biological antigens. Some of the known mechanisms by which SP-A contributes to innate immunity responses include a) opsonisation of specific bacteria for uptake by alveolar macrophages, b) recruitment of monocytes and neutrophils to the site of inflammation, c) enhancement of pathogen-killing mechanisms such as phagocytosis, and release of reactive oxygen intermediates, as well as nitric oxide, and d) modulation of cytokine production. Moreover, SP-A knockout mice are more susceptible to infection and show reduced pathogen clearance, phagocytosis by alveolar macrophages, and oxygen radical production [72]. In addition, SP-A may play a role in the transition of innate to adaptive immunity by its interaction with surface receptors of dendritic cells, an event that precedes antigen presentation to lymphocytes. Whether products of differential allele expression [73], and/or epigenetic modifications of SP-A genes [24] affect these and other innate or adaptive immunity processes remains to be determined. Known consequences of environmental insults in SP-A expression and function, as well as the recent discoveries of epigenetic mechanisms affecting the expression of the two SP-A genes and their variants by environmental challenges, are noted below.
Epigenetics of SP-A and environmental insults
Epigenetic mechanisms have been studied for the surfactant protein genes. We have summarised the major epigenetic modifications that affect SP-A gene expression in figure 1. Methylation of CpG sites located in the SP-A1 promoter is associated with lung cancer, and appears to correlate with changes in SP-A expression [24]. Preliminary studies from our group have also found associations of methylation of CpG sites in the SP-A2 promoter with lung cancer. Histone acetylation and methylation at regulatory regions of the SP-A gene promoters have been shown to affect SP-A expression in lung cells during development and under conditions such as hypoxia [74–76]. Moreover, a role of miRNAs in the regulation of SP-A expression has also been postulated [77], and alternative splicing at the SP-A 5’UTR is a major regulatory mechanism for differential SP-A1 and/or SP-A2 variant expression [77–79]. Although it remains to be determined whether epigenetic regulation of alternative splicing occurs at the 5’UTR of SP-A mRNAs, methylation of mRNA 5’UTRs holds the potential to regulate gene expression, by yet unknown mechanisms. In support of this, a recent study of 450,000 cytosine methylation sites in the human genome found approximately 200,000 CpG sites present in proximal promoters, and 25% of these were located at the 5’UTR [80].
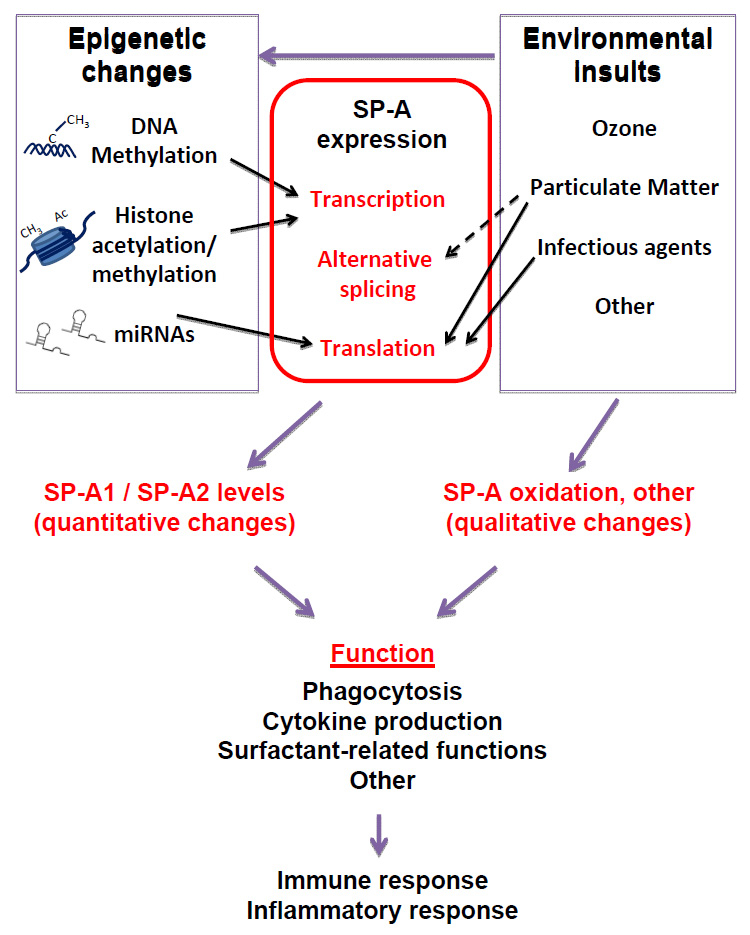
Figure 1
Epigenetics and the environment on SP-A expression and function. The effects of different environmental pollutants on epigenetics have been extensively studied and reported [1–6]. Environmental insults hold the potential to affect SP-A both qualitatively and quantitatively. Epigenetic mechanisms affect SP-A expression by altering transcription including methylation of CpG sites in the SP-A promoters [24, 74], and modifications of histones located in the chromatin of SP-A regulatory sequences [75, 76]), as well as translation efficiency (miRNA-mediated repression [77]). As a result, the relative content of SP-A1 and SP-A2 is affected, resulting in altered immune and inflammatory responses. SP-A is also affected by environmental insults. Oxidation of SP-A is increased after ozone exposure [43, 47], with subsequent effects on innate immunity and inflammatory processes and surfactant-related functions [26, 43, 85]. An effect of particulate matter on alternative splicing of SP-A transcripts (our unpublished data), and on translation may also occur [25]. Particulate matter and infection with RSV affect SP-A levels by altering translation efficiency [25, 83]. Arrows indicate published data; broken arrow indicates preliminary/unpublished data.
Oxidative stress caused by exposure to ozone affects the lower respiratory tract by causing cell damage and inflammation, as well as surfactant dysfunction [81]. Exposure to ozone significantly affects various SP-A functions, including its ability to modulate cytokine production [46, 47] and phagocytosis by alveolar macrophages [26]. Ozone exposure negatively affects mouse survival after bacterial infection, and ozone-induced oxidation of SP-A may negatively affect the survival outcome [82].
Environmental insults may also affect SP-A expression (fig. 1). In vitroexposure of lung cells to particulate matter affects the alternative splicing pattern of 5’UTR exons of SP-A1 transcripts (our preliminary studies), and SP-A translation, by activating cap-independent mechanisms [25]. Moreover, infectious agents such as respiratory syncytial virus affect SP-A translation efficiency [83]. We postulate that environmental exposures affect SP-A expression, both the total and the relative levels of SP-A1 and SP-A2. In fact, the latter has been correlated with aging and negative health outcomes [70, 84].
Concluding remarks
In the last few years, special attention has been given to the role of epigenetics in mediating not only genetic and physiological responses to certain environmental insults, but also in regulating underlying susceptibility to environmental stressors, as well as its effect on host defence. Molecules and cells involved in lung innate immunity, given their proximity to and contact with the external environment, are likely to be the first ones to be affected by environmental insults, via changes in their function and/or regulation. Such changes may initiate downstream effects and in turn may directly or indirectly modulate health outcomes. The example with the innate host defence molecule, SP-A, where epigenetic regulation differentially affects expression of its genetic and splice variants, points to the possibility that epigenetic mechanisms play a significant role in the underlying mechanisms of varied susceptibilities to a given disease.
Lung innate host defence molecules, and especially those found with extensive genetic complexity, such as SP-A, are likely to be key determinants of the differential downstream biological effects in response to environmental pollutants. Pollutants may differentially affect, via epigenetic changes or other mechanisms, the function and/or regulation of different variants of innate immunity. In turn, due to these differences in their regulation and/or function, innate immune molecules may differentially “set the ball rolling” as to who may be at disease risk and at what level of risk.
Future research investigating the impact of environmental pollutants on epigenetic regulation focusing on innate immunity molecules with natural genetic variability is likely to shed light on the underlying mechanisms of the basis of individual differences in disease susceptibility. The outcome of such studies is likely to benefit considerations of individualised medicine.
References
1 Herceg Z, Vaissière T. Epigenetic mechanisms and cancer: an interface between the environment and the genome. Epigenetics. 2011;6(7):804–19.
2 Hou L, Wang D, Baccarelli A. Environmental chemicals and microRNAs. Mutat Res. 2011;714(1–2):105–12.
3 Bollati V, Baccarelli A. Environmental epigenetics. Heredity. 2010;105(1):105–12.
4 Weidman JR, Dolinoy DC, Murphy SK, Jirtle RL. Cancer susceptibility: epigenetic manifestation of environmental exposures. Cancer J. 2007;13(1):9–16.
5 Faulk C, Dolinoy DC. Timing is everything: the when and how of environmentally induced changes in the epigenome of animals. Epigenetics. 2011;6(7):791–7.
6 Choudhuri S, Cui Y, Klaassen CD. Molecular targets of epigenetic regulation and effectors of environmental influences. Toxicol Appl Pharmacol. 2010;245(3):378–93.
7 Brunekreef B. The continuing challenge of air pollution. Eur Respir J. 2010;36(4):704–5.
8 Chen B, Kan H. Air pollution and population health: a global challenge. Environ Health Prev Med. 2008;13(2):94–101.
9 Lewtas J. Air pollution combustion emissions: characterization of causative agents and mechanisms associated with cancer, reproductive, and cardiovascular effects. Mutat Res. 2007;636(1–3):95–133.
10 Ciencewicki J, Trivedi S, Kleeberger SR. Oxidants and the pathogenesis of lung diseases. J Allergy Clin Immunol. 2008;122(3):456–68; quiz 469–70.
11 Romieu I, Moreno-Macias H, London SJ. Gene by environment interaction and ambient air pollution. Proc Am Thorac Soc. 2010;7(2):116–22.
12 Hassing C, Twickler M, Brunekreef B, Cassee F, Doevendans P, Kastelein J, et al. Particulate air pollution, coronary heart disease and individual risk assessment: a general overview. Eur J Cardiovasc Prev Rehabil. 2009;16(1):10–5.
13 Perez L, Rapp R, Künzli N. The Year of the Lung: outdoor air pollution and lung health. Swiss Med Wkly. 2010;140:w13129.
14 Jardim MJ. microRNAs: Implications for air pollution research. Mutat Res. 2011.
15 Bayarsaihan D. Epigenetic mechanisms in inflammation. J Dent Res. 2011;90(1):9–17.
16 Luco RF, Allo M, Schor IE, Kornblihtt AR, Misteli T. Epigenetics in alternative pre-mRNA splicing. Cell. 2011;144(1):16–26.
17 Kampa M, Castanas E. Human health effects of air pollution. Environ Pollut. 2008;151(2):362–7.
18 Baccarelli A, Wright RO, Bollati V, Tarantini L, Litonjua AA, Suh HH, et al. Rapid DNA methylation changes after exposure to traffic particles. Am J Respir Crit Care Med. 2009;179(7):572–8.
19 Floros J, Thomas N. Genetic variations of surfactant proteins and lung injury. Nakos G, Papathanasiou A (eds). In: Surfactant pathogenesis and treatment of lung disease. edn. Edited by Research Signpost K, India; 2009. p. 25–48.
20 Floros J, Pavlovic J. Genetics of acute respiratory distress syndrome: challenges, approaches, surfactant proteins as candidate genes. Semin Respir Crit Care Med. 2003;24(2):161–8.
21 Floros J, Phelps DS. Pulmonary surfactant. In: Yaksh TL, et al. (eds). Anesthesia: biologic foundations. Philadelphia: Lippincott-Raven Publishers; 1997. p. 1259–80.
22 Vaid M, Floros J. Surfactant protein DNA methylation: a new entrant in the field of lung cancer diagnostics? (Review). Oncol Rep. 2009;21(1):3–11.
23 Silveyra P, Floros J. Genetic variant associations of human SP-A and SP-D with acute and chronic lung injury. Front Biosci. 2012;17:407–29.
24 Lin Z, Thomas NJ, Bibikova M, Seifart C, Wang Y, Guo X, et al. DNA methylation markers of surfactant proteins in lung cancer. Int J Oncol. 2007;31(1):181–91.
25 Wang G, Guo X, Silveyra P, Kimball S, Floros J. Cap-independent translation of human SP-A 5'-UTR variants: a double-loop structure and cis-element contribution. Am J Physiol Lung Cell Mol Physiol. 2009;296(4):L635–47.
26 Mikerov A, Umstead T, Gan X, Huang W, Guo X, Wang G, et al. Impact of ozone exposure on the phagocytic activity of human surfactant protein A (SP-A) and SP-A variants. Am J Physiol Lung Cell Mol Physiol. 2008;294(1):L121–30.
27 Durrani F, Phelps DS, Weisz J, Silveyra P, Hu S, Mikerov AN, et al. Gonadal hormones and oxidative stress interaction differentially affects survival of male and female mice after lung Klebsiella Pneumoniae infection. Exp Lung Res. 2011;38(4):165–72.
28 Samet JM. The Clean Air Act and health – a clearer view from 2011. N Engl J Med. 2011;365(3):198–201.
29 Bernstein JA, Alexis N, Bacchus H, Bernstein IL, Fritz P, Horner E, et al. The health effects of non-industrial indoor air pollution. J Allergy Clin Immunol. 2008;121(3):585–91.
30 Ostro B, Lipsett M, Reynolds P, Goldberg D, Hertz A, Garcia C, et al. Long-term exposure to constituents of fine particulate air pollution and mortality: results from the California Teachers Study. Environ Health Perspect. 2010;118(3):363–9.
31 Guxens M, Sunyer J. A review of epidemiological studies on neuropsychological effects of air pollution. Swiss Med Wkly. 2012;141:w13322.
32 Bernstein JA, Alexis N, Barnes C, Bernstein IL, Nel A, Peden D, et al. Health effects of air pollution. J Allergy Clin Immunol. 2004;114(5):1116–23.
33 McCreanor J, Cullinan P, Nieuwenhuijsen MJ, Stewart-Evans J, Malliarou E, Jarup L, et al. Respiratory effects of exposure to diesel traffic in persons with asthma. N Engl J Med. 2007;357(23):2348–58.
34 Riedl MA. The effect of air pollution on asthma and allergy. Curr Allergy Asthma Rep. 2008;8(2):139–46.
35 Hollingsworth JW, Kleeberger SR, Foster WM. Ozone and pulmonary innate immunity. Proc Am Thorac Soc. 2007;4(3):240–6.
36 Smith KR, Jerrett M, Anderson HR, Burnett RT, Stone V, Derwent R, et al. Public health benefits of strategies to reduce greenhouse-gas emissions: health implications of short-lived greenhouse pollutants. Lancet. 2009;374(9707):2091–103.
37 Jerrett M, Burnett RT, Pope CA, 3rd, Ito K, Thurston G, Krewski D, et al. Long-term ozone exposure and mortality. N Engl J Med. 2009;360(11):1085–95.
38 Kim CS, Alexis NE, Rappold AG, Kehrl H, Hazucha MJ, Lay JC, et al. Lung function and inflammatory responses in healthy young adults exposed to 0.06 ppm ozone for 6.6 hours. Am J Respir Crit Care Med. 2011;183(9):1215–21.
39 Becker S, Madden MC, Newman SL, Devlin RB, Koren HS. Modulation of human alveolar macrophage properties by ozone exposure in vitro. Toxicol Appl Pharmacol. 1991;110(3):403–15.
40 Selgrade MK, Illing JW, Starnes DM, Stead AG, Menache MG, Stevens MA. Evaluation of effects of ozone exposure on influenza infection in mice using several indicators of susceptibility. Fundam Appl Toxicol. 1988;11(1):169–80.
41 Mikerov AN, Gan X, Umstead TM, Miller L, Chinchilli VM, Phelps DS, et al. Sex differences in the impact of ozone on survival and alveolar macrophage function of mice after Klebsiella pneumoniae infection. Respir Res. 2008;9:24.
42 Mikerov AN, Haque R, Gan X, Guo X, Phelps DS, Floros J. Ablation of SP-A has a negative impact on the susceptibility of mice to Klebsiella pneumoniae infection after ozone exposure: sex differences. Respir Res. 2008;9:77.
43 Haque R, Umstead TM, Ponnuru P, Guo X, Hawgood S, Phelps DS, et al. Role of surfactant protein-A (SP-A) in lung injury in response to acute ozone exposure of SP-A deficient mice. Toxicol Appl Pharmacol. 2007;220(1):72–82.
44 Janic B, Umstead TM, Phelps DS, Floros J. Modulatory effects of ozone on THP-1 cells in response to SP-A stimulation. Am J Physiol Lung Cell Mol Physiol. 2005;288(2):L317–25.
45 Gilmour MI, Hmieleski RR, Stafford EA, Jakab GJ. Suppression and recovery of the alveolar macrophage phagocytic system during continuous exposure to 0.5 ppm ozone. Exp Lung Res. 1991;17(3):547–58.
46 Wang G, Umstead TM, Phelps DS, Al-Mondhiry H, Floros J. The effect of ozone exposure on the ability of human surfactant protein a variants to stimulate cytokine production. Environ Health Perspect. 2002;110(1):79–84.
47 Huang W, Wang G, Phelps DS, Al-Mondhiry H, Floros J. Human SP-A genetic variants and bleomycin-induced cytokine production by THP-1 cells: effect of ozone-induced SP-A oxidation. Am J Physiol Lung Cell Mol Physiol. 2004;286(3):L546–53.
48 Durham AL, Wiegman C, Adcock IM. Epigenetics of asthma. Biochim Biophys Acta. 2011.
49 Madrigano J, Baccarelli A, Mittleman MA, Wright RO, Sparrow D, Vokonas PS, et al. Prolonged exposure to particulate pollution, genes associated with glutathione pathways, and DNA methylation in a cohort of older men. Environ Health Perspect. 2011;119(7):977–82.
50 Probst AV, Dunleavy E, Almouzni G. Epigenetic inheritance during the cell cycle. Nat Rev Mol Cell Biol. 2009;10(3):192–206.
51 Martienssen RA, Kloc A, Slotkin RK, Tanurdzić M. Epigenetic inheritance and reprogramming in plants and fission yeast. Cold Spring Harb Symp Quant Biol. 2008;73:265–71.
52 Nafee TM, Farrell WE, Carroll WD, Fryer AA, Ismail KM. Epigenetic control of fetal gene expression. BJOG. 2008;115(2):158–68.
53 Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006;31(2):89–97.
54 Shames DS, Minna JD, Gazdar AF. DNA methylation in health, disease, and cancer. Curr Mol Med. 2007;7(1):85–102.
55 Singh SM, Murphy B, O’Reilly RL. Involvement of gene-diet/drug interaction in DNA methylation and its contribution to complex diseases: from cancer to schizophrenia. Clin Genet. 2003;64(6):451–60.
56 Wen H, Schaller MA, Dou Y, Hogaboam CM, Kunkel SL. Dendritic cells at the interface of innate and acquired immunity: the role for epigenetic changes. J Leukoc Biol. 2008;83(3):439–46.
57 Pfefferle PI, Pinkenburg O, Renz H. Fetal epigenetic mechanisms and innate immunity in asthma. Curr Allergy Asthma Rep. 2010;10(6):434–43.
58 Awasthi S, Madhusoodhanan R, Wolf R. Surfactant protein-A and toll-like receptor-4 modulate immune functions of preterm baboon lung dendritic cell precursor cells. Cell Immunol. 2011;268(2):87–96.
59 Mikerov A, Umstead T, Huang W, Liu W, Phelps D, Floros J. SP-A1 and SP-A2 variants differentially enhance association of Pseudomonas aeruginosa with rat alveolar macrophages. Am J Physiol Lung Cell Mol Physiol. 2005;288(1):L150–8.
60 Crouch EC. Collectins and pulmonary host defense. Am J Respir Cell Mol Biol. 1998;19(2):177–201.
61 Crouch E, Hartshorn K, Ofek I. Collectins and pulmonary innate immunity. Immunol Rev. 2000;173:52–5.
62 Wright JR. Immunoregulatory functions of surfactant proteins. Nat Rev Immunol. 2005;5(1):58–68.
63 Mariencheck WI, Savov J, Dong Q, Tino MJ, Wright JR. Surfactant protein A enhances alveolar macrophage phagocytosis of a live, mucoid strain of P. aeruginosa. Am J Physiol. 1999;277(4 Pt 1):L777–86.
64 Wang J, Reid K. The immunoregulatory roles of lung surfactant collectins SP-A, and SP-D, in allergen-induced airway inflammation. Immunobiology. 2007;212(4-5):417–25.
65 Wang G, Phelps D, Umstead T, Floros J. Human SP-A protein variants derived from one or both genes stimulate TNF-alpha production in the THP-1 cell line. Am J Physiol Lung Cell Mol Physiol. 2000;278(5):L946–54.
66 Griese M, Birrer P, Demirsoy A. Pulmonary surfactant in cystic fibrosis. Eur Respir J. 1997;10(9):1983–8.
67 Hull J, South M, Phelan P, Grimwood K. Surfactant composition in infants and young children with cystic fibrosis. Am J Respir Crit Care Med. 1997;156(1):161–5.
68 Postle AD, Mander A, Reid KB, Wang JY, Wright SM, Moustaki M, et al. Deficient hydrophilic lung surfactant proteins A and D with normal surfactant phospholipid molecular species in cystic fibrosis. Am J Respir Cell Mol Biol. 1999;20(1):90–8.
69 Woodworth BA, Wood R, Baatz JE, Schlosser RJ. Sinonasal surfactant protein A1, A2, and D gene expression in cystic fibrosis: a preliminary report. Otolaryngol Head Neck Surg. 2007;137(1):34–8.
70 Tagaram H, Wang G, Umstead T, Mikerov A, Thomas N, Graff G, et al. Characterization of a human surfactant protein A1 (SP-A1) gene-specific antibody; SP-A1 content variation among individuals of varying age and pulmonary health. Am J Physiol Lung Cell Mol Physiol. 2007;292(5):L1052–63.
71 Mikerov A, Wang G, Umstead T, Zacharatos M, Thomas N, Phelps D, et al. Surfactant protein A2 (SP-A2) variants expressed in CHO cells stimulate phagocytosis of Pseudomonas aeruginosa more than do SP-A1 variants. Infect Immun. 2007;75(3):1403–12.
72 LeVine AM, Kurak KE, Wright JR, Watford WT, Bruno MD, Ross GF, et al. Surfactant protein-A binds group B streptococcus enhancing phagocytosis and clearance from lungs of surfactant protein-A-deficient mice. Am J Respir Cell Mol Biol. 1999;20(2):279–86.
73 Lin Z, Wang Y, Zhu K, Floros J. Differential allele expression of host defense genes, pulmonary surfactant protein-A and osteopontin, in rat. Mol Immunol. 2004;41(12):1155–65.
74 Benlhabib H, Mendelson CR. Epigenetic regulation of surfactant protein A gene (SP-A) expression in fetal lung reveals a critical role for Suv39h methyltransferases during development and hypoxia. Mol Cell Biol. 2011;31(10):1949–58.
75 Islam KN, Mendelson CR. Permissive effects of oxygen on cyclic AMP and interleukin-1 stimulation of surfactant protein A gene expression are mediated by epigenetic mechanisms. Mol Cell Biol. 2006;26(8):2901–12.
76 Islam KN, Mendelson CR. Glucocorticoid/glucocorticoid receptor inhibition of surfactant protein-A (SP-A) gene expression in lung type II cells is mediated by repressive changes in histone modification at the SP-A promoter. Mol Endocrinol. 2008;22(3):585–96.
77 Silveyra P, Wang G, Floros J. Human SP-A1 (SFTPA1) variant-specific 3' UTRs and poly(A) tail differentially affect the in vitro translation of a reporter gene. Am J Physiol Lung Cell Mol Physiol. 2010;299(4):L523–34.
78 Wang G, Guo X, Floros J. Differences in the translation efficiency and mRNA stability mediated by 5'-UTR splice variants of human SP-A1 and SP-A2 genes. Am J Physiol Lung Cell Mol Physiol. 2005;289(3):L497–508.
79 Silveyra P, Raval M, Simmons BP, Diangelo S, Wang G, Floros J. The untranslated exon B of human surfactant protein A2 (SFTPA2) mRNAs is an enhancer for transcription and translation. Am J Physiol Lung Cell Mol Physiol. 2011.
80 Sandoval J, Heyn HA, Moran S, Serra-Musach J, Pujana MA, Bibikova M, et al. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics. 2011;6(6):692–702.
81 Müller B, Seifart C, Barth PJ. Effect of air pollutants on the pulmonary surfactant system. Eur J Clin Invest. 1998;28(9):762–77.
82 Mikerov AN, Cooper TK, Wang G, Hu S, Umstead TM, Phelps DS, et al. Histopathologic evaluation of lung and extrapulmonary tissues show sex differences in Klebsiella pneumoniae – infected mice under different exposure conditions. Int J Physiol Pathophysiol Pharmacol. 2011;3(3):176–90.
83 Bruce SR, Atkins CL, Colasurdo GN, Alcorn JL. Respiratory syncytial virus infection alters surfactant protein A expression in human pulmonary epithelial cells by reducing translation efficiency. Am J Physiol Lung Cell Mol Physiol. 2009;297(4):L559–67.
84 Wang Y, Voelker DR, Lugogo NL, Wang G, Floros J, Ingram JL, et al. Surfactant protein-A is defective in abrogating inflammation in asthma. Am J Physiol Lung Cell Mol Physiol. 2011.
85 Wang G, Bates-Kenney S, Tao J, Phelps D, Floros J. Differences in biochemical properties and in biological function between human SP-A1 and SP-A2 variants, and the impact of ozone-induced oxidation. Biochemistry. 2004;43(14):4227–39.