Risky communication in atherosclerosis and thrombus formation
DOI: https://doi.org/10.4414/smw.2012.13553
Merlijn J. P. M. T
Meens, Anna
Pfenniger, Brenda R
Kwak
Summary
Atherosclerosis, a progressive disease of medium- and large-sized arteries, constitutes the major cause of death in developed countries, and is becoming increasingly prevalent in developing countries as well. The main consequences of atherosclerosis are myocardial infarction, cerebral infarction and aortic aneurysm. This inflammatory disease is characterised by specific intimal lesions where lipids, leukocytes and smooth muscle cells accumulate in the arterial wall over time. Risk factors for atherosclerosis can mainly be divided into two groups: i) risk factors induced by environment and behaviour (e.g., Western diet, smoking and sedentary lifestyle) and ii) genetic risk factors. Multiple epidemiological studies have associated a single nucleotide polymorphism (SNP) in the GJA4 gene, coding for connexin37 (Cx37), with increased risk for atherosclerosis and myocardial infarction. Connexins form gap junctions or hemi-channels that mediate an exchange of factors between i) the cytosol of two adjacent cells or ii) the cytosol and the extracellular environment, respectively. The GJA4 SNP codes for a proline-to-serine substitution at amino acid 319 in the regulatory C-terminus of the Cx37 protein, thereby altering basic and regulatory properties of its gap junction- and hemi-channels. In this review we discuss current evidence for mechanisms that link the GJA4SNP to atherosclerosis or thrombus formation after plaque rupture.
|
List of abbreviations
|
| ACS |
Acute coronary syndrome |
GSK-3 |
Glycogen synthase kinase-3 |
| ADP |
Adenosine diphosphate |
HIF |
Hypoxia-inducible factor |
| AMI |
Acute myocardial infarction |
IMT |
Intima-media thickness |
| ATP |
Adenosine triphosphate |
IP3 |
Inositol triphosphate |
| C1019T |
Cytosine-to-thymine substitution at position 1019 |
LDL |
Low-density lipoprotein |
| CAD |
Coronary artery disease |
LDLr-/-
|
LDL receptor deficient |
| cAMP |
Cyclic adenosine monophosphate |
MI |
Myocardial infarction |
| CL |
Cytoplasmic loop |
MMPs |
Matrix metalloproteases |
| CT |
COOH-terminus |
NO |
Nitric oxide |
| Cx |
Connexin |
NT |
NH2-terminus |
| ECs |
Endothelial cells |
oxLDL |
Oxidised low density lipoprotein |
| EL |
Extracellular loop |
PAD |
Peripheral artery disease |
| ELs |
Extracellular loops |
PDGF |
Platelet-derived growth factor |
| eNOS |
Endothelial nitric oxide synthase |
SMCs |
Smooth muscle cells |
| FMD |
Flow-mediated dilatation |
SNP |
Single nucleotide polymorphism |
| GJA4 |
Gap junction protein alpha 4 gene |
TRAP-6 |
Thrombin receptor activator for peptide 6 |
| GJIC |
Gap junctional intercellular communication |
|
|
Atherosclerosis
The main consequences of atherosclerosis, an inflammatory disease of medium- and large-sized arteries, are myocardial infarction (MI), stroke, peripheral artery disease (PAD) and aortic aneurysm. This disease causes more morbidity and mortality in the Western world than any other disorder [1]. Moreover, since the Western way of life is quickly spreading to other parts of the world, atherosclerosis-related diseases are currently rapidly spreading to the rest of the world as well. Therefore, atherosclerosis is a major threat to human health worldwide. As a consequence, the atherosclerotic process has been studied extensively using both samples from humans as well as animals models. These studies have revealed that atherogenesis, the process of plaque development, follows distinct phases during which specific cells have their own roles (see [2–7] for excellent reviews). The disease is generally considered as a chronic inflammatory and healing response to endothelial cell injury [6, 8–10]. The specific pathways contributing to endothelial injury or dysfunction are not completely understood but include well-known risk factors for the disease such as hypertension, hypercholesterolemia, toxins from cigarette smoke, homocysteine and hemodynamic factors [5, 11–13]. Moreover, atherosclerotic lesions are not randomly localised along the arterial tree. They tend to occur at dividing points and ostia of branches exiting vessels where disturbed blood flow patterns occur [14, 15]. Dysfunctional endothelial cells (ECs) show increased endothelial permeability, enhanced leukocyte and platelet adhesion, and altered gene expression [5, 16]. Low-density lipoprotein (LDL) accumulates within the intima, the sub-endothelial layer of the vascular wall. LDL is subsequently oxidised through the action of reactive oxygen species that are generated by ECs or macrophages in the intima. The presence of oxidised LDL (oxLDL) is implicated in plaque development via several pathways: i) oxLDL enhances expression of adhesion molecules, chemotactic proteins and growth factors by ECs, resulting in recruitment of monocytes and other inflammatory cells [5]; ii) the presence of oxLDL inhibits production of athero-protective nitric oxide (NO) [17], thereby reducing vasodilator activity; iii) oxLDL is ingested by macrophages (present in the initial lesion due to activation of the attracted monocytes) through the scavenger receptor and accumulates in these phagocytes, thus transforming them to foam cells (i.e. macrophages containing large lipid droplets) [5]. Foam cells secrete pro-inflammatory cytokines and thus further contribute to the development of an inflammatory environment and leukocyte recruitment in the initial lesion [5]. Over time, foam cells die due to apoptosis and necrosis leaving behind a mass of cellular debris in the centre of the plaque called the necrotic core [5]. In addition to the secretion of pro-inflammatory cytokines, macrophages, activated ECs and locally adherent platelets secrete growth factors, such as platelet-derived growth factor (PDGF), which cause migration of vascular smooth muscle cells (SMCs) towards the intima [2, 5, 18, 19]. Initially, these SMCs cover the plaque and give rise to the fibrous cap through secretion of extracellular matrix, most notably collagen [20]. Therefore, attraction of SMCs is important for plaque stabilisation. However, as the lesion progresses, enzymes (e.g. matrix metalloproteases (MMPs)) secreted by activated inflammatory cells start to degrade the extracellular matrix deposited by the SMCs [20]. Thinning of the fibrous cap results in an unstable plaque prone to rupture [20, 21]. Most clinical events related to atherosclerosis, such as myocardial and cerebral infarction, are caused by the rupture of the cap, which exposes the highly thrombogenic material in the plaque to the blood stream [21]. This induces the formation of a thrombus, which might occlude the vessel and cause ischemia in the tissue irrigated by it. In conclusion, atherosclerosis is a complex disease involving crosstalk between various cells and molecules. It is therefore of vital importance to understand the cellular and molecular mechanisms underlying the risk factors involved in the development of this pathology. Risk factors of atherosclerosis are typically subdivided into modifiable (environmental) ones, such as hyperlipidemia, hypertension, cigarette smoking, diabetes, sedentary lifestyle and C-reactive protein, and non-modifiable factors, like increasing age, male gender and family history [13]. Although a Mendelian genetic disorder, such as familial hypercholesterolemia, has been found in a small percentage of cases, the familial predisposition is multi-factorial relating to inheritance of a multitude of genetic polymorphisms [22, 23]. In recent years, many epidemiological studies have linked genetic polymorphisms in at least 100 human genes to increased risk for atherosclerosis using different clinical endpoints [23]. Mechanistic understanding for some of these gene polymorphisms has been obtained from additional studies on knockout mice. In this review we discuss current insights regarding the role of one of these genetic risk factors, a gene polymorphism for connexin37 (Cx37), in plaque development and in thrombus formation, the ultimate consequence of plaque rupture.
Connexins, connexons and gap junctions
Connexins are a family of trans-membrane proteins with 20 members in mice and 21 in humans (see [24] for a review regarding connexin regulation and function). Connexin genes were cloned for the first time in 1986 [25]. They are relatively simple and consist of a 5’-untranslated exon, an intron of varying length, an exon that contains the uninterrupted coding region and a 3’-untranslated region. The nomenclature used to indicate connexin genes is based on similarities between gene sequences and the length of the cytoplasmic loop [26]. It separates the connexins into four groups: alpha, beta, gamma and delta. In this system Cx37 is called alpha4 (GJA4), since it is the fourth connexin of the alpha group to be identified [26]. The nomenclature of connexin proteins is different; they are named according to their predicted molecular weight [26]. Thus, Cx37 has a predicted molecular weight of around 37 kD. Connexin proteins consist of four trans-membrane helices, two extracellular loops (ELs), a cytoplasmic loop (CL) and cytoplasmic NH2- and COOH-termini (NT and CT) [27].

Figure 1
Connexins form connexons composed of six connexins in the Golgi apparatus. Thereafter, connexons can be inserted in the plasma membrane where they can function as connexons (hemi-channels; left) or as gap junctions (right) if they dock to a connexon expressed by a neighbouring cell. Connexons allow exchange of small molecules (<1000 Da) between the cytosol and the extracellular space. Gap junctions allow passage of similar molecules between adjacent cells.
Connexins do not function as single proteins. Six connexins assemble into connexons, also called hemi-channels, and two connexons from neighbouring cells dock in the extracellular space to form a full gap junction channel (fig. 1) [24]. Connexons can consist of one type of connexin (homomeric connexon) or of different connexins (heteromeric connexon). Connexins enter as monomers in the endoplasmic reticulum (e.g., Cx43 [28]), they oligomerise in the Golgi apparatus and then traffic along microtubules towards the plasma membrane [29, 30]. During intracellular trafficking, the connexon generally remains closed to avoid exchange of diffusible factors between the cytosolic and intra-vesicular compartments. Moreover, connexons generally remain closed after insertion into the plasma membrane [24], but they can switch to an open state depending on environmental factors (e.g., the intracellular and extracellular Ca2+ concentration [24], presence of arginine etc.). Such open hemi-channels allow passive exchange of small molecules (<1000 Da; e.g., adenosine triphosphate (ATP), Ca2+, cAMP and IP3) through their hydrophilic pore between the cytosol and the extracellular space. These exchanges have physiological roles; they are important for cell volume regulation, paracrine and autocrine signalling [24, 31].
More classically, connexins are important for mediating exchange of cytosolic factors between two adjacent cells. This exchange can occur when two hemi-channels dock via a non-covalent mechanism and form a gap junction channel (fig. 1). Whether two connexons can dock is mainly determined by the properties of the ELs [24, 32–35]. Gap junctions are agglomerations of multiple (sometimes thousands) gap junction channels and their typical appearance with electron microscopy was first recognised and described in 1967 [36]. Again, a gap junction channel can consist of two identical connexons or of two different connexons. The former gap junction channel is called homotypic while the latter type is called heterotypic. Gap junctions are found in almost every tissue and are involved in various processes, such as cellular growth, proliferation and differentiation, synchronisation of contractions in the heart and spreading of vasodilation in different vascular beds [24].
Many groups have used in vitro studies with transfection of communication-deficient tumour cell lines or expression in oocytes to characterise properties of channels formed by individual connexins (see for example [37] and [38]). As mentioned above, it appeared that the highly conserved ELs mainly regulate docking properties [24, 32–35]. Interestingly, patch clamp measurements revealed that gap junction channels or hemi-channels formed by different connexins display different biophysical characteristics in terms of unitary conductance, open probability and permeability [37, 39]. Thus, hemi-channels consisting of Cx37 may allow other molecules to pass compared to hemi-channels consisting of Cx43. In contrast to the EL regions, the CL and CT are variable in length and composition. These domains regulate the activity of connexin channels and its responses to changes in its environment. CT-mediated regulation of channel properties frequently involves changes in the phosphorylation status of the CT by various kinases [40, 41]. In addition, the CT interacts with several binding partners (e.g., calmodulin, eNOS and tight junction proteins [42–45]). Such interactions of the CT with other proteins may also have consequences for channel activity [42, 45]. In addition to acute regulation of channel activity, the phosphorylation status of the CT has been proposed to be one of the regulators of gap junction assembly and removal from the plasma membrane [46]. Indeed, connexins have a relatively short half-life ranging from 1 to 5 hours [30].
Knockout mice have also been used extensively to study the function of connexins in vivo[43, 47–49]. However, interpretation of these studies is not always simple since connexins have overlapping functions, and thus expression of a particular connexin may compensate for loss of another [50] or may not cause a phenotype because the basal expression of other connexins is sufficient to maintain normal connexin function. For instance, knockout of a connexin (e.g., Cx40) was shown to increase expression of Cx37 [51] in endothelial cells. However, in a more recent study, knockout of Cx40 caused decreased expression of Cx37 in the endothelium but increased expression in the media [43]. Finally, endothelial-specific knockout of Cx40 decreased the expression of Cx37 in these cells [52]. Thus, a general consensus regarding the effect of absence of Cx40 on Cx37 has not been reached and will require more work. Finally, it has been shown that, in some cases, knockout of a connexin may lead to a reduced expression of another connexion [43, 53].
Expression of connexins during atherosclerosis
Four connexins are expressed in the vasculature. In healthy arteries, Cx37 and Cx40 are mainly expressed by ECs where both are in a complex with eNOS, suggesting that they may have a role in NO-mediated endothelium-dependent vasodilatation [43, 45, 54]. In addition, it has been shown that Cx40 is required for the spread of endothelium-dependent vasodilatation in small arteries, such as mouse arterioles [55]. Some ECs at branching points of arteries, regions submitted to a highly turbulent flow, have been shown to over-express Cx43 in addition to Cx37 and Cx40 [56]. SMCs in the arterial media express Cx43 and Cx45. Interestingly, studies on the atherosclerosis model of LDL receptor-deficient (LDLr–/–) mice showed that the expression of connexins in large arteries like the aorta changes during atherosclerosis (fig. 2, table 1 and [54]). Moreover, plaques at different stages (e.g., early fatty streak or advanced atheroma) are characterised by a specific pattern of connexin expression [54]. In early atheromas ECs express Cx37 and Cx40. In addition, macrophage-derived foam cells present express Cx37. Cx43 expression in intimal SMCs is up-regulated. In advanced atheromas, connexin expression has changed even more. ECs covering an advanced atheroma no longer express connexins, with the exception of ECs at the shoulder region that express Cx43. Foam cells close to the necrotic core express both Cx37 and Cx43, and Cx43 expression in intimal SMCs in down-regulated. SMCs in the medial layer of the artery beneath an advanced atheroma start to express Cx37. As described before, each connexin forms gap junction- and hemi-channels with different conductance, permeability and regulation properties. Therefore, the change in connexin expression pattern might differentially regulate the communication between the different atheroma-associated cell types involved.
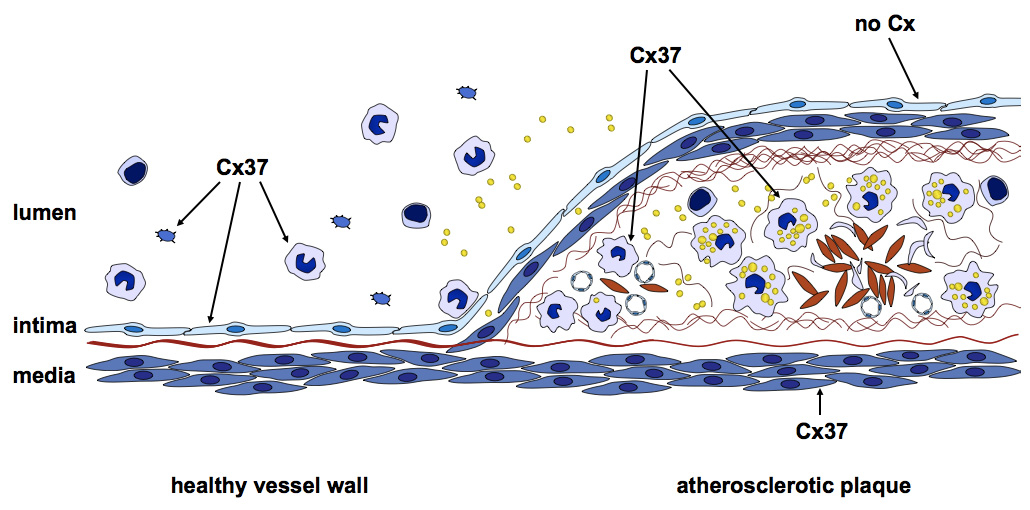
Figure 2
Connexins are differentially expressed during atherosclerotic plaque development. In the healthy vessel wall (left), Cx37 is expressed by ECs and monocytes. In contrast, at the site of an atherosclerotic plaque (right), Cx37 is expressed by SMCs and macrophages and is no longer expressed by the endothelium overlaying the lesion.
|
Table 1: Expression of connexons during atherosclerotic plaque development. ND: not determined. |
| |
Healthy vessel
|
Early atheroma
|
Advanced atheroma
|
| Endothelial cells |
Cx37, Cx40 |
Cx37, Cx40 |
Cx43 (only in shoulder region). |
| Monocytes / macrophages |
Cx37 |
Cx37 |
Cx37, Cx43 (in cells close to lipid core) |
| Smooth muscle cells (intima) |
– |
Cx43 |
Cx43 |
| Smooth muscle cells (media) |
Cx43 |
Cx43 |
Cx37, Cx43 |
| Platelets |
Cx37 |
ND |
ND |
Cx37: a prognostic marker in disease?
Mutations and polymorphisms in genes coding for connexins have been associated with various pathologies such as several types of cancer, deafness and lymphedema (see [57] for a recent review). Mutated Cx37 was first described to be a tumour-associated antigen in murine Lewis lung carcinoma cell lines [58]. Subsequent studies in various human lung and breast cancers revealed polymorphisms rather than mutations in the GJA4gene [59–61]. Krutovskikh and colleagues in 1996 described a first GJA4 gene polymorphism that resulted in an amino acid change (valine-to-isoleucine) at codon 130 situated in the CL of the Cx37 protein [61]. Subsequently, a second interesting polymorphism corresponding to a cytosine-to-thymine substitution at position 1019 (C1019T) was discovered in the human population [62]. This single nucleotide polymorphism (SNP) results in a non-conservative proline-to-serine amino acid change in the CT of Cx37, and Cx37-319P was shown to be over-represented in individuals exhibiting significant atherosclerotic plaques in the common carotid artery in a Swedish population [62]. Being located in the regulatory CT of a connexin protein, it was immediately speculated that this SNP could have significant impact on channel function. On the one hand, the serine residue in Cx37-319S is part of a serine-rich area of the protein and predicted to be subject of phosphorylation by glycogen synthase kinase-3 (GSK-3). On the other hand, proline residues, like the one in Cx37-319P, are known to cause bends in polypeptide chains. Consequently, loss of this amino acid may affect the tertiary structure of the Cx37 C-terminal domain and its possibility to interact with other proteins. Indeed, detailed patch clamp studies on transfected cells revealed functional differences between human Cx37 polymorphic gap junction- and hemi-channels under basal conditions [63]. Surprisingly, subsequent studies identified a unique site in Cx37-319P for phosphorylation by GSK-3. Phosphorylation of this serine 321 site in Cx37-319P was associated with reduced gap junctional intercellular communication (GJIC), whereas GJIC remained unaffected by similar treatment in Cx37-319S expressing cells [64], suggesting that the regulatory properties of those two types of gap junction channels are also different. Since the original study by Boerma and colleagues [62], the Cx37-C1019T polymorphism has been shown to associate with carotid intima-media thickness (IMT), coronary artery disease (CAD), acute myocardial infarction (AMI) or MI in several but not all populations investigated (table 2) [65–76]. Moreover, data suggests that this polymorphism can be used as a prognostic marker for carotid IMT, ACS, MI or ischemic stroke (table 2) [62, 77, 78]. Interestingly, in young Finns, a population in which the Cx37 SNP is not directly associated with carotid IMT, CAD or flow-mediated dilatation (FMD), it has been shown that this polymorphism affects other cardiovascular risk factors such as smoking and homocysteine [12]. Furthermore, the C1019T SNP has been associated with PAD in type 2 diabetics suggesting that it may play a role in the peripheral vasculature as well [79].
|
Table 2: Overview of studies on possible associations between the GJA4 SNP and diseases involving, at least to some extent, atherosclerosis. |
|
Disease / surrogate marker
|
Association / prognostic factor
|
Population
|
Study size
|
Ref
|
| Carotid IMT |
C allele |
Swedish males |
1155 subjects |
[62] |
| CAD |
T allele |
Taiwanese males / females |
177 patients, 102 controls |
[72] |
| MI |
T allele |
Japanese males / females |
2819 patients, 2242 controls |
[71] |
| CAD |
T allele |
Japanese males / females |
3085 patients, 2122 controls |
[70] |
| CAD |
T allele |
Japanese males / females |
1011 patients, 650 controls |
[94] |
| AMI |
T allele |
Sicilian males |
97 patients, 196 controls |
[68] |
| CAD / MI |
C allele |
Swiss males / females |
597 patients, 184 controls |
[78] |
| CAD |
No association / prognostic factor |
Irish males / females |
416 patients, 490 sibling controls |
[74] |
| Carotid IMT, carotid artery compliance, brachial artery FMD |
No association / prognostic factor |
Finnish males / females |
1440 individuals |
[73] |
| CAD |
C allele |
Northern Han Chinese males / females |
502 patients, 410 controls |
[65] |
| MI |
T allele |
Sicilian males |
97 patients, 196 controls |
[67] |
| Acute coronary syndrome (ACS) |
T allele |
American males / females |
695 ACS patients |
[77] |
| Ankle brachial blood pressure index |
T allele |
Japanese males / females |
2288 type 2 diabetics |
[79] |
| ACS |
No association / prognostic factor |
Czech males / females |
1686 ACS patients |
[75] |
| Ankle brachial blood pressure index |
T allele |
Czech females |
178 type 1 diabetics, 111 type 2 diabetics, 862 controls |
[69] |
| Carotid IMT, ischemic stroke |
T allele |
Taiwanese males / females |
3330 subjects |
[66] |
| Ischemic stroke |
No association / prognostic factor |
Taiwanese males / females |
958 patients, 2196 controls |
[76] |
Role of Cx37 in plaque development
Since many of the pathologies for which Cx37 is a prognostic marker are caused by atherosclerosis, it has been hypothesised that Cx37 has a role in the atherosclerotic process and that this role is influenced by the different Cx37 polymorphisms. This hypothesis has recently been tested using Cx37-deficient animals on an atherosclerosis-prone ApoE–/– background [49]. On a high cholesterol diet, Cx37-deficient mice developed atherosclerosis more rapidly compared to Cx37-expressing ApoE–/–littermates [49]. Interestingly, in vivo adoptive transfer experiments, in which macrophages extracted from a mouse are injected into another mouse, showed that deletion of Cx37 from macrophages but not from ECs caused enhanced recruitment to atherosclerotic lesions, suggesting that Cx37 has a role in monocyte adhesion. These results were confirmed on a limited number of mice using monocytes instead of macrophages, suggesting that Cx37 affects monocyte transmigration into the intima [49]. Subsequently in vitro studies on isolated monocytes and macrophages demonstrated that the presence or absence of Cx37 modulates adhesion, one of the early steps of leukocyte recruitment. As the experiments were designed on individual monocytes and no gap junctions were formed between them, it suggested that hemi-channels might play a role in the regulation of cell adhesion. ATP has been shown to inhibit monocyte adhesion [80, 81] and to pass through gap junctions [82, 83]. Therefore, a link between Cx37 in monocytes/macrophages and ATP secretion was investigated. Indeed, Cx37-expressing monocytes/macrophages released more ATP in the extracellular environment than Cx37-deficient cells. In addition, when extracellular ATP was degraded by the enzyme apyrase, increased adhesion of Cx37-expressing monocytes/macrophages was observed [49]. Altogether, these experiments suggested that Cx37 mediates a decrease in leukocyte adhesion through an autocrine ATP-dependent mechanism. Of note, these macrophages also displayed increased ATP- and Cx37-dependent adhesion to endothelial cells in culture [49]. As aforementioned, the C1019T polymorphism has functional consequences and is a prognostic factor for several pathologies that are at least partly caused by atherosclerosis [62, 63, 65–68, 71, 72, 77, 78]. It was therefore hypothesised that the Cx37 SNP polymorphism may also affect monocyte adhesion. Indeed, expression of Cx37-319S or Cx37-319P in a communication deficient monocyte cell line resulted in decreased cell adhesion due to increased ATP secretion [49]. Interestingly, cell adhesion was even more reduced by expression of Cx37-319P compared to expression of Cx37-319S [49]. Therefore, individuals carrying Cx37-319P may be protected against early atherosclerosis due to decreased monocyte adhesion. Hence, the C1019T polymorphism may be used as a prognostic biomarker to screen for individuals at risk for plaque development.
Role of Cx37 in platelets
Rapid formation of a blood clot is required to avoid excessive loss of blood after vascular injury. However, rapid thrombus formation may be a disadvantage after plaque rupture. Thus, individuals displaying decreased blood clot formation after vascular injury, like individuals treated with platelet inhibitors, may be better protected against acute events during which a thrombus or multiple thrombi cause blood vessel occlusion and ischemia. Thrombus formation involves three phases; initiation, extension and perpetuation. Each of these stages requires synchronised actions of multiple cells and thus a fine-tuned intercellular communication. This suggests that platelets, blood cells involved in blood clotting, may display communication via gap junctions during this process. To assess whether platelets express connexins, we recently performed immunofluorescence microscopy and Western blots on protein isolated from resting and activated platelets [47]. These experiments showed that platelets express Cx37 but not Cx40 or Cx43 [47]. Expression of Cx37 was not confined to platelets alone since Cx37 expression was also observed in their precursors, megakaryocytes [47].
Mice lacking Cx37 displayed decreased bleeding time in a tail transection model [47] suggesting that Cx37 is involved in either blood clotting or the vasomotor response induced by vascular injury. However, ex vivovasomotor responses in Cx37-deficient mice were unaltered compared to wild-type mice. In addition, Cx37-deficient mice displayed enhanced thrombo-embolism after injection of a mixture of collagen and epinephrine in the jugular vein [47] suggesting that the prothrombotic phenotype of these mice might be due to changes in platelet aggregation. This was further investigated using platelet aggregation assays performed on platelets isolated from Cx37-deficient and control mice. Indeed, irrespective of the agonist used (i.e., arachidonic acid, ADP, collagen or thrombin), aggregation of platelets isolated from Cx37-deficient mice was enhanced compared to aggregation of platelets isolated from control mice [47]. In another experiment, platelets isolated from wild-type mice were used to study the effect of blockage of gap junctions on platelet aggregation. In line with the aggregation assays performed on the platelets isolated from Cx37-deficient mice, inhibition of gap junctions using a connexin-mimetic peptide caused increased platelet aggregation compared to wild-type platelets treated with a control peptide [47]. Interestingly, this was also the case for aggregation of human platelets using another pharmacological connexin inhibitor (alpha-glycyrrethinic acid) [47]. In the latter experiment it was noted that platelets isolated from some individuals (in this case 3 out of 6) display more pronounced aggregation in response to collagen, arachidonic acid or TRAP-6 suggesting that the Cx37 SNP may be involved in platelet aggregation responses. Thus, 96 healthy male volunteers of Caucasian descent participating in a Swiss platelet function study were genotyped for the Cx37 polymorphism. Indeed, subjects homozygous for GJA4-1019C, coding for Cx37-319P, displayed increased platelet responses compared with individuals carrying the GJA4-1019T allele [47]. Although the study awaits further confirmation in a larger population, the results indicate that the Cx37 SNP may constitute a marker for platelet reactivity. Finally, Cx37-319P gap junction channels displayed decreased permeability for neurobiotin, a Cx37-permeant tracer molecule [47]. Altogether, these data provided the first evidence that the establishment of GJIC between Cx37-expressing platelets may be a mechanism to limit thrombus propensity. The authors propose a model in which an anti-aggregating signal, such as cAMP, may spread more efficiently between platelets through Cx37-319S gap junction channels, thereby limiting exaggerated accumulation of platelets. However, it should be kept in mind that the data are also consistent with a model in which decrease of GJIC between Cx37-319P platelets enhances the intracellular concentration of pro-aggregating signals in the newly recruited platelets at the edge of the thrombus resulting in further recruitment.
Future perspectives
In conclusion, studies on knockout mice have revealed that Cx37 is atheroprotective. Presence of Cx37 hemi-channels in monocytes decreases ATP-dependent monocyte adhesion, resulting in a reduced build-up of plaques [49] and presumably more stable lesions [84]. Cx37-319S polymorphic hemi-channels are less permeable for ATP than Cx37-319P hemi-channels, which leads to stronger monocyte adhesion [49, 63]. Thus, Cx37-319P could function as a protective genetic variant by specifically delaying the recruitment of monocytes to atherosclerotic lesions. Another important finding from studies on knockout mice is that communication through Cx37 gap junction channels limits thrombus propensity [47]. Cx37-319P gap junction channels display decreased intercellular communication [64] resulting in augmented platelet aggregation responses in Cx37-319P bearing individuals. These 2 mechanistic results onto the Cx37 SNP might seem in conflict with each another at first sight if one would consider that the risk for MI is a simple sum of the number of macrophages in the atherosclerotic lesion and thrombosis following plaque rupture. The vulnerability of an atherosclerotic lesion is indeed strongly determined by its inflammatory cell component, but additional factors play a role as well. Recently, increasing evidence points to intraplaque haemorrhage as a critical factor in promoting atherosclerotic lesion instability. Vasa vasorum, a network of microvasculature originating primarily in the adventitia of large arteries, become activated during atherosclerosis in human and mice [85]. Micro-vessel content is known to increase with plaque progression, a process that is likely stimulated by plaque hypoxia, reactive oxygen species and hypoxia-inducible factor (HIF) signalling [86]. However, plaque micro-vessels are immature and fragile and the distorted integrity of the micro-vessel endothelium likely leads to intraplaque haemorrhage. The associated accumulation of red blood cell-derived cholesterol and rapid expansion of the necrotic core as well as the influx of macrophages involved in red blood cell and iron phagocytosis places the plaque at increased risk for rupture [86–88]. Human atherosclerotic lesions have also been shown to contain lymphatic vessels [86] and, similar to blood micro-vessels, lymphatic micro-vessel content also increases with plaque progression [89]. The role of lymphatic vessels in atherosclerosis is at present unclear. Interestingly, Cx37 has been shown to regulate arteriogenesis, angiogenesis and vasculogenesis in a mouse model of post-ischemic hindlimb recovery [90, 91]. Moreover, failure of lymphatic valve formation and lymph drainage has been recently reported in Cx37-deficient mice [92, 93]. Future studies on mice with cell-specific deletion of Cx37 should reveal the relative importance of Cx37 in monocytes/macrophages, platelets, blood ECs and lymphatic ECs with respect to plaque stability. It would be premature to design pharmacological strategies to specifically target Cx37 hemi-channels in monocytes or gap junction channels in platelets until additional insight has been obtained on this matter.
It is more realistic is to believe that the GJA4 SNP may in the near future be used as a prognostic marker for myocardial infarction or recurrence of events for example. Obviously, multi-factorial diseases are influenced by differential expression of various genes and screening patients for one marker only may not be very useful. However, it may be worthwhile to consider the use of this SNP in combination with the other polymorphisms associated with atherosclerosis [23] to screen which prognostic marker(s) are present in a given patient. Ultimately, such a genetic profiling would allow clinicians to, at least in theory, adapt therapy to the need of the individual patient and obviously, in the current era of personalised medicine, this seems a promising prospect.
Acknowledgements: We thank Cindy Wong, Laurent Burnier, Marc Chanson, Pierre Fontana and Anne Angelillo-Scherrer for helpful discussions.
References
1 Roger VL, Go AS, Lloyd-Jones DM, Adams RJ, Berry JD, Brown TM, et al. Heart disease and stroke statistics – 2011 update: a report from the American Heart Association. Circulation. 2011;123(4):e18–e209.
2 Libby P. Inflammation in atherosclerosis. Nature. 2002;420(6917):868–74.
3 Libby P, Aikawa M. Stabilization of atherosclerotic plaques: new mechanisms and clinical targets. Nat Med. 2002;8(11):1257–62.
4 Libby P, DiCarli M, Weissleder R. The vascular biology of atherosclerosis and imaging targets. J Nucl Med. 2010;51(Suppl 1):33S–37S.
5 Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473(7347):317–25.
6 Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol. 2011;12(3):204–12.
7 Shibata N, Glass CK. Regulation of macrophage function in inflammation and atherosclerosis. J Lipid Res. 2009;50(Suppl):S277–81.
8 Ross R. Atherosclerosis – an inflammatory disease. N Engl J Med. 1999;340(2):115–26.
9 Libby P, Okamoto Y, Rocha VZ, Folco E. Inflammation in atherosclerosis: transition from theory to practice. Circ J. 2010;74(2):213–20.
10 Davignon J, Ganz P. Role of endothelial dysfunction in atherosclerosis. Circulation. 2004;109(23 Suppl 1):III27–32.
11 Bornfeldt KE, Tabas I. Insulin resistance, hyperglycemia, and atherosclerosis. Cell Metab. 2011;14(5):575–85.
12 Collings A, Raitakari OT, Juonala M, Mansikkaniemi K, Kahonen M, Hutri-Kahonen N, et al. The influence of smoking and homocysteine on subclinical atherosclerosis is modified by the connexin37 C1019T polymorphism – The Cardiovascular Risk in Young Finns Study. Clin Chem Lab Med. 2008;46(8):1102–8.
13 Smith SC, Jr., Milani RV, Arnett DK, Crouse JR, 3rd, McDermott MM, Ridker PM, et al. Atherosclerotic Vascular Disease Conference: Writing Group II: risk factors. Circulation. 2004;109(21):2613–6.
14 Davies PF. Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nat Clin Pract Cardiovasc Med. 2009;6(1):16–26.
15 Davies PF, Spaan JA, Krams R. Shear stress biology of the endothelium. Ann Biomed Eng. 2005;33(12):1714–8.
16 Flammer AJ, Luscher TF. Three decades of endothelium research: from the detection of nitric oxide to the everyday implementation of endothelial function measurements in cardiovascular diseases. Swiss Med Wkly. 2010;140:w13122.
17 Cominacini L, Rigoni A, Pasini AF, Garbin U, Davoli A, Campagnola M, et al. The binding of oxidized low density lipoprotein (ox-LDL) to ox-LDL receptor-1 reduces the intracellular concentration of nitric oxide in endothelial cells through an increased production of superoxide. J Biol Chem. 2001;276(17):13750–5.
18 Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84(3):767–801.
19 Sano H, Sudo T, Yokode M, Murayama T, Kataoka H, Takakura N, et al. Functional blockade of platelet-derived growth factor receptor-beta but not of receptor-alpha prevents vascular smooth muscle cell accumulation in fibrous cap lesions in apolipoprotein E-deficient mice. Circulation. 2001;103(24):2955–60.
20 Newby AC, Zaltsman AB. Fibrous cap formation or destruction – the critical importance of vascular smooth muscle cell proliferation, migration and matrix formation. Cardiovasc Res. 1999;41(2):345–60.
21 Yla-Herttuala S, Bentzon JF, Daemen M, Falk E, Garcia-Garcia HM, Herrmann J, et al. Stabilisation of atherosclerotic plaques. Position paper of the European Society of Cardiology (ESC) Working Group on atherosclerosis and vascular biology. Thromb Haemost. 2011;106(1):1–19.
22 Soutar AK, Naoumova RP. Mechanisms of disease: genetic causes of familial hypercholesterolemia. Nat Clin Pract Cardiovasc Med. 2007;4(4):214–25.
23 Stylianou IM, Bauer RC, Reilly MP, Rader DJ. Genetic basis of atherosclerosis: insights from mice and humans. Circ Res. 2012;110(2):337–55.
24 Saez JC, Berthoud VM, Branes MC, Martinez AD, Beyer EC. Plasma membrane channels formed by connexins: their regulation and functions. Physiol Rev. 2003;83(4):1359–400.
25 Kumar NM, Gilula NB. Cloning and characterization of human and rat liver cDNAs coding for a gap junction protein. J Cell Biol. 1986;103(3):767–76.
26 Sohl G, Willecke K. An update on connexin genes and their nomenclature in mouse and man. Cell Commun Adhes. 2003;10(4-6):173–80.
27 Unger VM, Kumar NM, Gilula NB, Yeager M. Three-dimensional structure of a recombinant gap junction membrane channel. Science. 1999;283(5405):1176–80.
28 Musil LS, Goodenough DA. Biochemical analysis of connexin43 intracellular transport, phosphorylation, and assembly into gap junctional plaques. J Cell Biol. 1991;115(5):1357–74.
29 Martin PE, Evans WH. Incorporation of connexins into plasma membranes and gap junctions. Cardiovasc Res. 2004;62(2):378–87.
30 Laird DW. Life cycle of connexins in health and disease. Biochem J. 2006;394(Pt 3):527–43.
31 Bosco D, Haefliger JA, Meda P. Connexins: key mediators of endocrine function. Physiol Rev. 2011;91(4):1393–445.
32 White TW, Paul DL, Goodenough DA, Bruzzone R. Functional analysis of selective interactions among rodent connexins. Mol Biol Cell. 1995;6(4):459–70.
33 White TW, Bruzzone R, Wolfram S, Paul DL, Goodenough DA. Selective interactions among the multiple connexin proteins expressed in the vertebrate lens: the second extracellular domain is a determinant of compatibility between connexins. J Cell Biol. 1994;125(4):879–92.
34 Dahl G, Werner R, Levine E, Rabadan-Diehl C. Mutational analysis of gap junction formation. Biophys J. 1992;62(1):172–80; discussion 180–2.
35 Dahl G, Levine E, Rabadan-Diehl C, Werner R. Cell/cell channel formation involves disulfide exchange. Eur J Biochem. 1991;197(1):141–4.
36 Revel JP, Karnovsky MJ. Hexagonal array of subunits in intercellular junctions of the mouse heart and liver. J Cell Biol. 1967;33(3):C7–C12.
37 Kwak BR, Hermans MM, De Jonge HR, Lohmann SM, Jongsma HJ, Chanson M. Differential regulation of distinct types of gap junction channels by similar phosphorylating conditions. Mol Biol Cell. 1995;6(12):1707–19.
38 Morley GE, Taffet SM, Delmar M. Intramolecular interactions mediate pH regulation of connexin43 channels. Biophys J. 1996;70(3):1294–302.
39 Moreno AP. Biophysical properties of homomeric and heteromultimeric channels formed by cardiac connexins. Cardiovasc Res. 2004;62(2):276–86.
40 Lampe PD, Lau AF. The effects of connexin phosphorylation on gap junctional communication. Int J Biochem Cell Biol. 2004;36(7):1171–86.
41 Kwak BR, van Veen TA, Analbers LJ, Jongsma HJ. TPA increases conductance but decreases permeability in neonatal rat cardiomyocyte gap junction channels. Exp Cell Res. 1995;220(2):456–63.
42 Herve JC, Bourmeyster N, Sarrouilhe D. Diversity in protein-protein interactions of connexins: emerging roles. Biochim Biophys Acta. 2004;1662(1-2):22–41.
43 Alonso F, Boittin FX, Beny JL, Haefliger JA. Loss of connexin40 is associated with decreased endothelium-dependent relaxations and eNOS levels in the mouse aorta. Am J Physiol Heart Circ Physiol. 2010;299(5):H1365–73.
44 Looft-Wilson RC, Billaud M, Johnstone SR, Straub AC, Isakson BE. Interaction between nitric oxide signaling and gap junctions: Effects on vascular function. Biochim Biophys Acta. 2011.
45 Pfenniger A, Derouette JP, Verma V, Lin X, Foglia B, Coombs W, et al. Gap junction protein Cx37 interacts with endothelial nitric oxide synthase in endothelial cells. Arterioscler Thromb Vasc Biol. 2010;30(4):827–34.
46 Solan JL, Lampe PD. Connexin phosphorylation as a regulatory event linked to gap junction channel assembly. Biochim Biophys Acta. 2005;1711(2):154–63.
47 Angelillo-Scherrer A, Fontana P, Burnier L, Roth I, Sugamele R, Brisset A, et al. Connexin 37 limits thrombus propensity by downregulating platelet reactivity. Circulation. 2011;124(8):930–9.
48 Odermatt B, Wellershaus K, Wallraff A, Seifert G, Degen J, Euwens C, et al. Connexin 47 (Cx47)-deficient mice with enhanced green fluorescent protein reporter gene reveal predominant oligodendrocytic expression of Cx47 and display vacuolized myelin in the CNS. J Neurosci. 2003;23(11):4549–59.
49 Wong CW, Christen T, Roth I, Chadjichristos CE, Derouette JP, Foglia BF, et al. Connexin37 protects against atherosclerosis by regulating monocyte adhesion. Nat Med. 2006;12(8):950–4.
50 Lopez D, Rodriguez-Sinovas A, Agullo E, Garcia A, Sanchez JA, Garcia-Dorado D. Replacement of connexin 43 by connexin 32 in a knock-in mice model attenuates aortic endothelium-derived hyperpolarizing factor-mediated relaxation. Exp Physiol. 2009;94(10):1088–97.
51 Kruger O, Beny JL, Chabaud F, Traub O, Theis M, Brix K, et al. Altered dye diffusion and upregulation of connexin37 in mouse aortic endothelium deficient in connexin40. J Vasc Res. 2002;39(2):160–72.
52 Chadjichristos CE, Scheckenbach KE, van Veen TA, Richani Sarieddine MZ, de Wit C, Yang Z, et al. Endothelial-specific deletion of connexin40 promotes atherosclerosis by increasing CD73-dependent leukocyte adhesion. Circulation. 2010;121(1):123–31.
53 Simon AM, McWhorter AR. Decreased intercellular dye-transfer and downregulation of non-ablated connexins in aortic endothelium deficient in connexin37 or connexin40. J Cell Sci. 2003;116(Pt 11):2223–36.
54 Kwak BR, Mulhaupt F, Veillard N, Gros DB, Mach F. Altered pattern of vascular connexin expression in atherosclerotic plaques. Arterioscler Thromb Vasc Biol. 2002;22(2):225–30.
55 Wolfle SE, Schmidt VJ, Hoepfl B, Gebert A, Alcolea S, Gros D, et al. Connexin45 cannot replace the function of connexin40 in conducting endothelium-dependent dilations along arterioles. Circ Res. 2007;101(12):1292–9.
56 Gabriels JE, Paul DL. Connexin43 is highly localized to sites of disturbed flow in rat aortic endothelium but connexin37 and connexin40 are more uniformly distributed. Circ Res. 1998;83(6):636–43.
57 Pfenniger A, Wohlwend A, Kwak BR. Mutations in connexin genes and disease. Eur J Clin Invest. 2011;41(1):103–16.
58 Mandelboim O, Berke G, Fridkin M, Feldman M, Eisenstein M, Eisenbach L. CTL induction by a tumour-associated antigen octapeptide derived from a murine lung carcinoma. Nature. 1994;369(6475):67–71.
59 Saito T, Barbin A, Omori Y, Yamasaki H. Connexin 37 mutations in rat hepatic angiosarcomas induced by vinyl chloride. Cancer Res. 1997;57(3):375–7.
60 Saito T, Krutovskikh V, Marion MJ, Ishak KG, Bennett WP, Yamasaki H. Human hemangiosarcomas have a common polymorphism but no mutations in the connexin37 gene. Int J Cancer. 2000;86(1):67–70.
61 Krutovskikh V, Mironov N, Yamasaki H. Human connexin 37 is polymorphic but not mutated in tumours. Carcinogenesis. 1996;17(8):1761–3.
62 Boerma M, Forsberg L, Van Zeijl L, Morgenstern R, De Faire U, Lemne C, et al. A genetic polymorphism in connexin 37 as a prognostic marker for atherosclerotic plaque development. J Intern Med. 1999;246(2):211–8.
63 Derouette JP, Desplantez T, Wong CW, Roth I, Kwak BR, Weingart R. Functional differences between human Cx37 polymorphic hemichannels. J Mol Cell Cardiol. 2009;46(4):499–507.
64 Morel S, Burnier L, Roatti A, Chassot A, Roth I, Sutter E, et al. Unexpected role for the human Cx37 C1019T polymorphism in tumour cell proliferation. Carcinogenesis. 2010;31(11):1922–31.
65 Han Y, Xi S, Zhang X, Yan C, Yang Y, Kang J. Association of connexin 37 gene polymorphisms with risk of coronary artery disease in northern Han Chinese. Cardiology. 2008;110(4):260–5.
66 Leu HB, Chung CM, Chuang SY, Bai CH, Chen JR, Chen JW, et al. Genetic variants of connexin37 are associated with carotid intima-medial thickness and future onset of ischemic stroke. Atherosclerosis. 2011;214(1):101–6.
67 Listi F, Candore G, Balistreri CR, Caruso M, Incalcaterra E, Hoffmann E, et al. Connexin37 1019 gene polymorphism in myocardial infarction patients and centenarians. Atherosclerosis. 2007;191(2):460–1.
68 Listi F, Candore G, Lio D, Russo M, Colonna-Romano G, Caruso M, et al. Association between C1019T polymorphism of connexin37 and acute myocardial infarction: a study in patients from Sicily. Int J Cardiol. 2005;102(2):269–71.
69 Pitha J, Hubacek JA, Pithova P. The connexin 37 (1019C>T) gene polymorphism is associated with subclinical atherosclerosis in women with type 1 and 2 diabetes and in women with central obesity. Physiol Res. 2010;59(6):1029–32.
70 Yamada Y, Ichihara S, Izawa H, Tanaka M, Yokota M. Genetic risk for coronary artery disease in individuals with or without type 2 diabetes. Mol Genet Metab. 2004;81(4):282–90.
71 Yamada Y, Izawa H, Ichihara S, Takatsu F, Ishihara H, Hirayama H, et al. Prediction of the risk of myocardial infarction from polymorphisms in candidate genes. N Engl J Med. 2002;347(24):1916–23.
72 Yeh HI, Chou Y, Liu HF, Chang SC, Tsai CH, Connexin37 gene polymorphism and coronary artery disease in Taiwan. Int J Cardiol. 2001;81(2-3):251–5.
73 Collings A, Islam MS, Juonala M, Rontu R, Kahonen M, Hutri-Kahonen N, et al. Associations between connexin37 gene polymorphism and markers of subclinical atherosclerosis: the Cardiovascular Risk in Young Finns study. Atherosclerosis. 2007;195(2):379–84.
74 Horan PG, Allen AR, Patterson CC, Spence MS, McGlinchey PG, McKeown PP. The connexin 37 gene polymorphism and coronary artery disease in Ireland. Heart. 2006;92(3):395–6.
75 Hubacek JA, Stanek V, Gebauerova M, Pilipcincova A, Poledne R, Aschermann M, et al. Lack of an association between connexin-37, stromelysin-1, plasminogen activator-inhibitor type 1 and lymphotoxin-alpha genes and acute coronary syndrome in Czech Caucasians. Exp Clin Cardiol. 2010;15(3):e52–6.
76 Juo SH, Liao YC, Lin HF, Chen PL, Lin WY, Lin RT. Lack of association between a functional genetic variant of connexin 37 and ischemic stroke in a Taiwanese population. Thromb Res. 2012.
77 Lanfear DE, Jones PG, Marsh S, Cresci S, Spertus JA, McLeod HL. Connexin37 (GJA4) genotype predicts survival after an acute coronary syndrome. Am Heart J. 2007;154(3):561–6.
78 Wong CW, Christen T, Pfenniger A, James RW, Kwak BR. Do allelic variants of the connexin37 1019 gene polymorphism differentially predict for coronary artery disease and myocardial infarction? Atherosclerosis. 2007;191(2):355–61.
79 Katakami N, Sakamoto K, Kaneto H, Matsuhisa M, Shimizu I, Ishibashi F, et al. Association between the connexin37 polymorphism and peripheral arterial disease in subjects with type 2 diabetes. Diabetes Care. 2009;32(5):e53–4.
80 Henttinen T, Jalkanen S, Yegutkin GG. Adherent leukocytes prevent adenosine formation and impair endothelial barrier function by Ecto-5'-nucleotidase/CD73-dependent mechanism. J Biol Chem. 2003;278(27):24888–95.
81 Kunapuli SP, Daniel JL. P2 receptor subtypes in the cardiovascular system. Biochem J. 1998;336(Pt 3):513–23.
82 Goldberg GS, Lampe PD, Nicholson BJ. Selective transfer of endogenous metabolites through gap junctions composed of different connexins. Nat Cell Biol. 1999;1(7):457–9.
83 Leybaert L, Braet K, Vandamme W, Cabooter L, Martin PE, Evans WH. Connexin channels, connexin mimetic peptides and ATP release. Cell Commun Adhes. 2003;10(4-6):251–7.
84 Derouette JP, Wong C, Burnier L, Morel S, Sutter E, Galan K, et al. Molecular role of Cx37 in advanced atherosclerosis: a micro-array study. Atherosclerosis. 2009;206(1):69–76.
85 Virmani R, Kolodgie FD, Burke AP, Finn AV, Gold HK, Tulenko TN, et al. Atherosclerotic plaque progression and vulnerability to rupture: angiogenesis as a source of intraplaque hemorrhage. Arterioscler Thromb Vasc Biol. 2005;25(10):2054–61.
86 Sluimer JC, Daemen MJ. Novel concepts in atherogenesis: angiogenesis and hypoxia in atherosclerosis. J Pathol. 2009;218(1):7–29.
87 Kolodgie FD, Narula J, Yuan C, Burke AP, Finn AV, Virmani R. Elimination of neoangiogenesis for plaque stabilization: is there a role for local drug therapy? J Am Coll Cardiol. 2007;49(21):2093–101.
88 Michel JB, Virmani R, Arbustini E, Pasterkamp G. Intraplaque haemorrhages as the trigger of plaque vulnerability. Eur Heart J. 2011;32(16):1977–85, 1985a, 1985b, 1985c.
89 Kholova I, Dragneva G, Cermakova P, Laidinen S, Kaskenpaa N, Hazes T, et al. Lymphatic vasculature is increased in heart valves, ischaemic and inflamed hearts and in cholesterol-rich and calcified atherosclerotic lesions. Eur J Clin Invest. 2011;41(5):487–97.
90 Fang JS, Angelov SN, Simon AM, Burt JM. Cx37 deletion enhances vascular growth and facilitates ischemic limb recovery. Am J Physiol Heart Circ Physiol. 2011;301(5):H1872–81.
91 Fang JS, Angelov SN, Simon AM, Burt JM. Cx40 is required for, and cx37 limits, postischemic hindlimb perfusion, survival and recovery. J Vasc Res. 2012;49(1):2–12.
92 Kanady JD, Dellinger MT, Munger SJ, Witte MH, Simon AM. Connexin37 and Connexin43 deficiencies in mice disrupt lymphatic valve development and result in lymphatic disorders including lymphedema and chylothorax. Dev Biol. 2011;354(2):253–66.
93 Sabine A, Agalarov Y, Maby-El Hajjami H, Jaquet M, Hagerling R, Pollmann C, et al. Mechanotransduction, PROX1, and FOXC2 Cooperate to Control Connexin37 and Calcineurin during Lymphatic-Valve Formation. Dev Cell, 2012.
94 Hirashiki A, Yamada Y, Murase Y, Suzuki Y, Kataoka H, Morimoto Y, et al. Association of gene polymorphisms with coronary artery disease in low- or high-risk subjects defined by conventional risk factors. J Am Coll Cardiol. 2003;42(8):1429–37.