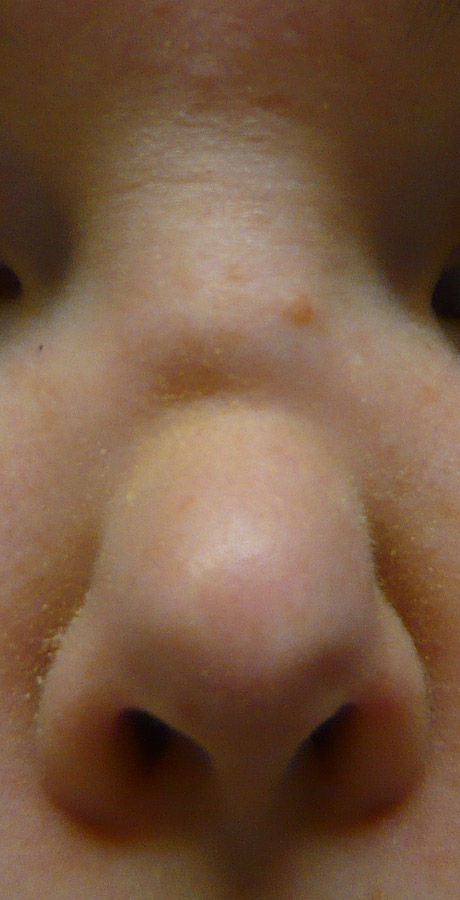
Figure 1
Nasal deformity in a patient with granulomatosis with polyangiitis (GPA). (Written informed consent for publication has been obtained from the patient.)
DOI: https://doi.org/10.4414/smw.2012.13541
1 Lecture delivered at the Annual meeting of the Swiss Respiratory Society in Interlaken (4–6 May 2011).
Systemic vasculitides are rare and potentially life-threatening diseases, especially if the diagnosis is delayed and appropriate treatment is not promptly initiated. Granulomatosis with polyangiitis (GPA; formerly Wegener’s granulomatosis) and Churg Strauss syndrome (possibly to be renamed eosinophilic granulomatosis with polyangiitis; EGPA) are the two main systemic vasculitides, both mainly affecting small-sized vessels, that may involve the upper respiratory tract (URT) [1–6]. Chronic allergic rhinitis and nasal polyposis are frequent in EGPA and recurrent chronic sinusitis and/or otitis in GPA, and are often the initial but non-specific manifestations, before other vasculitis symptoms [4, 7]. Although not absolutely specific, severe nasal crusting, nasal septal perforation, saddle nose deformity and/or subglottic stenosis (SGS) are more suggestive of GPA [8–10]. Awareness of these diseases as potential causes of URT manifestations is prerequisite for prompt diagnosis and initiation of appropriate treatment. Vasculitis outside the ears, nose and/or throat (ENT) usually responds well to systemic therapy, but upper airway manifestations tend to be relatively more refractory and/or to linger [11]. Thus, local treatments are a regular important component of therapeutic management for patients with URT manifestations, along with close collaboration with specialised otorhinolaryngologists.
Vasculitis is defined histologically as inflammation within the wall of the blood vessels, resulting in occlusion and/or thrombosis of vessel lumen and, ultimately, tissue ischaemia and/or organ injury. This inflammation can be granulomatous and/or associated with fibrinoid necrosis of the vessel wall. Classification of systemic vasculitides is mainly based on histological features and the size of the vessel predominantly involved, from large- to medium- and finally small-sized vessel vasculitides according to the Chapel Hill nomenclature [12]. GPA, EGPA and microscopic polyangiitis (MPA), the main three small-sized vessel vasculitides, are associated with serum anti-neutrophil cytoplasm antibodies (ANCA) in some 80%, 40% and 70% of patients respectively [6, 8, 13–16]. Other yet unidentified factors and pathogenic mechanisms may explain the different clinical patterns of the known vasculitides, such as more frequent ENT involvement in GPA than in MPA or giant cell arteritis. A recent hypothesis is the endothelial expression of different toll-like receptors depending on the vessel and its size [17–19]. ENT infections may be involved in the pathogenesis of GPA, at least as potential factors triggering disease flares and/or contributing to chronic inflammation in nasal and sinus mucosa [20–23]. Evidence of a role for infections in the pathogenesis of EGPA and MPA is weaker. Repeated environmental exposure to silica, cattle, solvents and/or dust is associated with increased risk of ANCA-associated vasculitis but is observed in less than 10% of patients in practice [24–26].
GPA prevalence is about 50 to 100 per million inhabitants in European countries [27]. The main target organs are the ENT and URT (85–95%); the kidneys, with pauci-immune glomerulonephritis (40–70%); and the lungs (40–60%), with alveolar haemorrhage of variable degrees and/or parenchymal nodules, which can be excavated. Other frequent clinical manifestations include purpuric infiltrated skin lesions, which are sometimes necrotic, and peripheral nerve involvement, mainly mononeuritis multiplex. Most GPA patients show involvement of several organs at diagnosis, along with some constitutional symptoms such as fever, weight loss, arthralgias or myalgias; however, the disease may initially remain limited, especially to the ENT and/or lungs, in up to 30% of patients [4, 13, 28–31]. Persistently localised GPA is rarer, accounting for less than 5% of all GPA cases after 5 years or more of follow-up in both French and German cohorts [8, 32]. The diagnosis in such patients with localised, limited, non-severe, early systemic or non-systemic GPA (depending on the definition used) can be challenging. More than 90% of patients with systemic GPA but only 60% to 80% with a limited form have cytoplasmic-ANCA C-ANCA, with a diffuse cytoplasmic labelling pattern on immunofluorescence (IF) assay and directed towards proteinase 3 (PR3) on enzyme-linked immunosorbent assay (ELISA). Although highly suggestive of GPA, anti-PR3 C-ANCA positivity is not entirely specific. Positivity for perinuclear-ANCA (P-ANCA; perinuclear labelling pattern on IF assay) directed towards myeloperoxidase (MPO) on ELISA is rare but possible. Evidence of (granulomatous) vasculitis on biopsy can be helpful to support a GPA diagnosis.
Figure 1
Nasal deformity in a patient with granulomatosis with polyangiitis (GPA). (Written informed consent for publication has been obtained from the patient.)
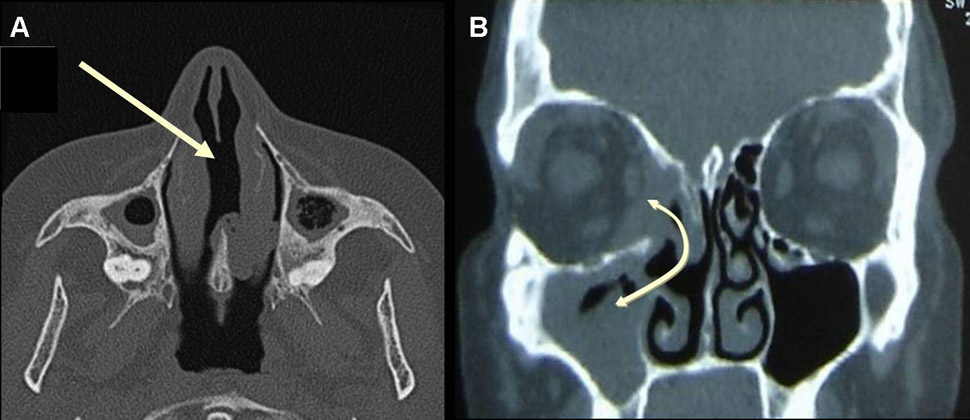
Figure 2
A) CT scan of the sinus in a patient with GPA, showing nasal septum perforation (arrow) and maxillary sinusitis with some degree of maxillary sinusitis and atrophy, with osteosclerosis and bony thickening of the paranasal sinuses. B) CT scan of the sinus in a patient with GPA showing sinusitis and sinus wall erosion leading to the formation of a communication (fistula) between right maxillary sinus, nasal cavity and orbit (double-headed arrow).
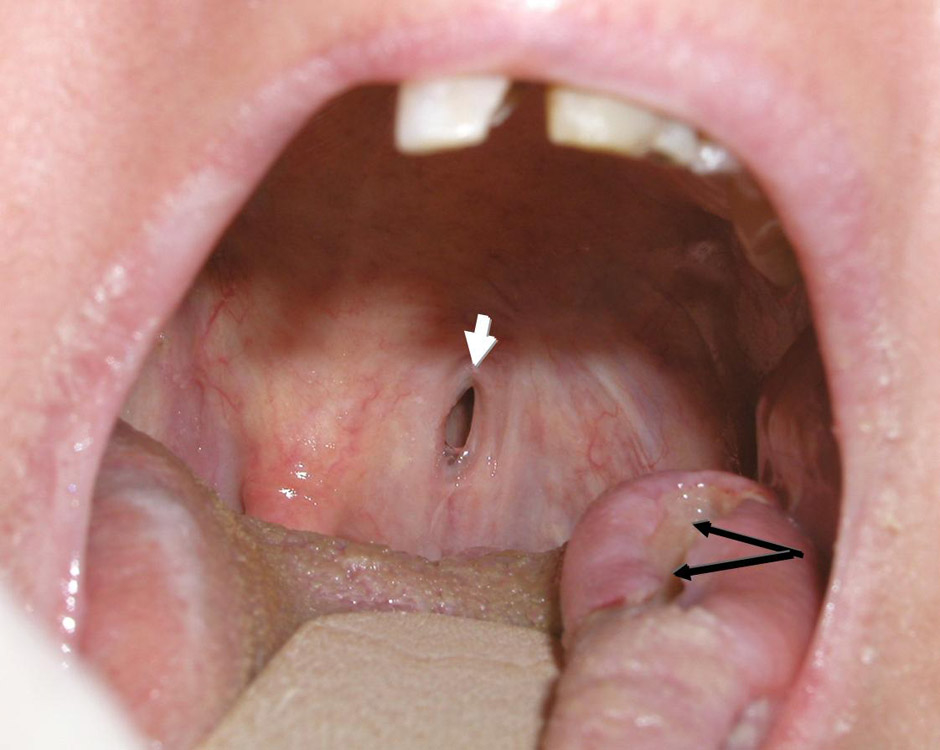
Figure 3
Lateral tongue ulceration (black arrows) and palatal fistula (white arrow) in a patient with GPA.
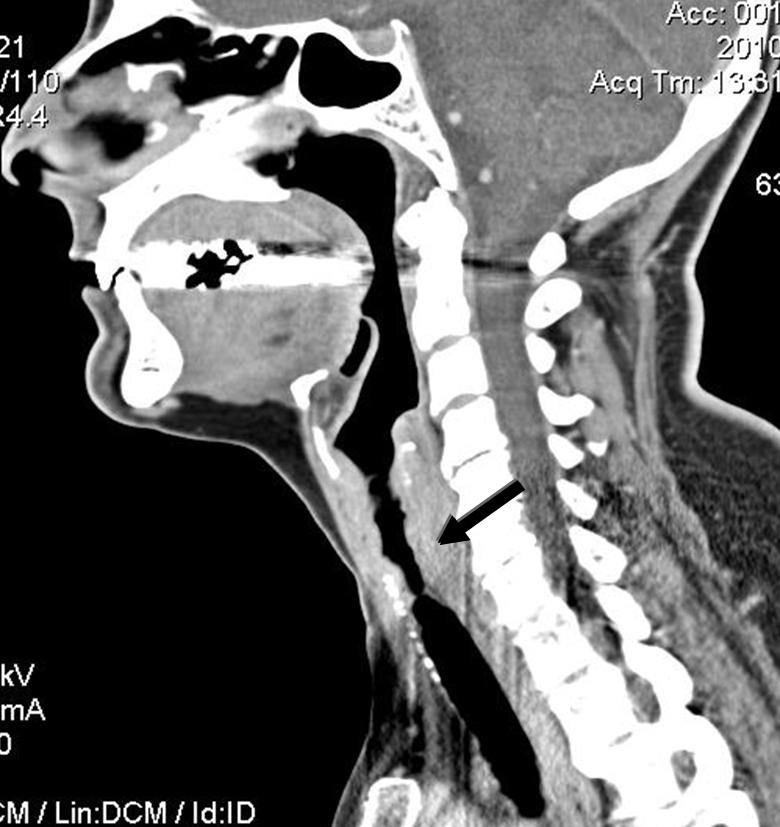
Figure 4
CT scan of the neck of a patient with GPA showing 6-cm-long subglottic stenosis (arrow).
Sino-nasal involvement is the most common manifestation of GPA in the head and neck, occurring in up to 85% of patients [33, 34]. Rhinitis is almost always present, with nasal dripping, obstruction, congestion, crusting and often epistaxis. Sinusitis, with sinus pain, is frequently erosive and/or leads to sinus atrophy over time. Anosmia or hyposmia is a frequent complaint of GPA patients [35]. The nasal and sinus mucosa usually appear inflamed on endoscopy, often ulcerated and occasionally with a granulomatous or “cobblestone” appearance. On CT scan of sinuses, mucosal thickening in the nasal cavity and the paranasal sinuses is the most frequent finding, especially in the maxillary sinuses, along with total or subtotal opacification of the paranasal sinuses and/or the mastoid cells. Bony erosion is present in more than half of patients with sino-nasal involvement and sclerosing osteitis of the paranasal sinuses and/or the mastoid cells in up to 20% [10].
Nasal cartilage erosion and lysis are classic signs of GPA, although not totally specific, and can result in nasal septum perforation, saddle nose deformity (fig. 1 and 2A) and/or orbito-sinusal or sinuso-palatal fistula (fig. 2B and 3). Nasal septum perforation usually occurs in the anterior nasal septum in the area of Kiesselbach’s plexus and is suggestive of GPA but not specific. Cocaine-induced perforation, infection (as in tuberculosis), and antiphospholipid syndrome are other potential causes. However, in contrast to GPA, septal perforation related to cocaine abuse is usually associated with otherwise clean URT mucosa. Saddle nose deformity occurs in 10% to 25% of patients, quite rapidly and chiefly during the early phases of the disease; however, it is also not pathognomonic for GPA. Other causes include bare lymphocyte syndrome (deficit in transporter for antigen presentation), relapsing polychondritis, congenital syphilis, paranasal sinus malignancy, midline lymphoma or, simply, trauma. Why some patients develop nasal deformity or septum perforation and others do not remains unexplained. To date, studies have only demonstrated more frequent nasal deformity with SGS (on univariable analysis) and retro-orbital masses (on uni- and multivariable analyses) [36].
Serous otitis media with effusion is the most common otological manifestation of GPA, present in up to one-third of patients [37]. It is thought to be secondary to Eustachian tube dysfunction resulting from physical obstruction by granulomatous involvement of the Eustachian tube or nasopharynx. It is often recurrent and/or chronic, possibly evolving to mastoiditis and/or requiring the insertion of transtympanic aeration tubes that tend to become rapidly obstructed or ejected. Granulomatous lesions of the middle ear are possible but infrequent. Conductive hearing loss may occur as a result of middle ear effusion. Sensorineural hearing loss can affect up to one-third of patients and may occur rapidly, progressing over days to weeks [10, 34]. The pathogenesis of sensorineural hearing loss may relate to cochlear vascular inflammation or deposition of immune complexes within the cochlea [34, 37]. Facial nerve palsy has been reported in up to 8–10% of patients with ENT involvement [37].
Tongue or gum ulcerations are not infrequent in GPA (fig. 3), as is gum sensitivity or hypertrophic strawberry-like gingivitis [38]. In addition to lingual ulcerations, infarction of the tongue has been reported.
The infra-glottic trachea is considered anatomically to be part of the lower respiratory tract. However, SGS, a rare but extremely challenging manifestation of GPA, is best managed by otorhinolaryngologists in collaboration with internists or rheumatologists for adjuvant systemic therapy. This manifestation was initially reported as affecting as many as 23% of patients. In more recent registry or cohort studies, such as the French, German or North American ones, the frequency is close to 3% to 9%. SGS occurs more frequently in women (female:male ratio 6:1) and in patients younger than average (i.e., approx. age 35 rather than 50) [32, 36, 39]. It is also a frequent manifestation in children with GPA. SGS can be isolated as the presenting or, rarely, the only manifestation of GPA [40]. The tracheal lumen can become significantly narrowed, a development which can be life-threatening. Obstruction can occur secondarily to acute inflammation due to suprainfection or detached crusts or scabs. Such cases may require urgent tracheotomy. To prevent this situation, physicians need a high level of suspicion and SGS must be included in the differential diagnosis for patients with GPA and dyspnoea or voice changes [41]. Such patients should be promptly evaluated by an otorhinolaryngologist. SGS may occasionally be associated with bronchial stenoses, which are even rarer than SGS but also potentially life-threatening, and in the majority of cases managed by respirologists.
SGS is best studied on CT scan (fig. 4 and 5) but must also be evaluated clinically by flexible nasolaryngoscopy. Interestingly, the lesion is usually segmental and located 1 to 2 cm below the vocal cords, probably because it represents the junction between 2 embryological buds or growth centres. Biopsy of the lesions can be revealing in more than half of cases, but endoscopy and biopsy of SGS may be risky in such patients. These invasive procedures can trigger a local inflammatory reaction, worsen the stenosis, and/or result in detached necrotic debris which is then inhaled, during the procedure itself or soon after. Spirometry may reveal blunting of the inspiratory curve, but the diagnosis should not rely solely on this modality.
EGPA is rarer than GPA, with a prevalence of about 10 to 15 per million inhabitants in the countries of Europe [27]. In the classic form patients present with long-lasting pseudo-allergic polyposis and/or with ENT manifestations in almost half of cases, and/or late-onset asthma, eosinophilia and eosinophilic tissue infiltration, especially in the lungs. Lung infiltrates are usually transient and labile, and inspection of broncho-alveolar lavage fluid reveals eosinophil-rich content. Up to one-third of EGPA patients are positive for P-ANCA on IF assay, usually with anti-MPO specificity on ELISA, ENT manifestations are notably more frequent in ANCA-positive patients whereas cardiac manifestations, associated with poor survival, are more frequent in ANCA-negative patients.
Pseudo-allergic rhinitis, sinusitis and nasal polyps are the most common ENT manifestations in EGPA [42]. Apart from anecdotal cases, such nasal and sinus lesions are typically not erosive in EGPA as compared with GPA. However, crusting and epistaxis are possible in EGPA patients, as is anosmia or hyposmia [35]. Polyps and less frequently soft-tissue masses within the sinus and/or, rarely, the orbits may cause obstruction, infiltrate through the anatomical orifices of the skull and/or have a mass effect, leading to compression of adjacent structures such as nerves. These lesions will almost always recur after surgery and in the absence of combined systemic treatment.
The utility of ENT mucosa biopsy for diagnostic purposes in GPA or EGPA with sino-nasal or mouth lesions remains disputed. Sensitivity of systematic nasal mucosa biopsy is low. Vasculitis or granuloma is seen in less than 10% of such biopsies. However, the probability of detecting histological features supporting a diagnosis of GPA or EGPA increases by up to 50% with major lesions in sino-nasal mucosa from which a deep biopsy can be taken (i.e. with the patient usually under general anaesthesia or intravenous sedation) [4, 43, 44]. The potential for airway obstruction and respiratory compromise must be considered before proceeding with biopsy of the URT mucosa.
Management of vasculitis patients with URT manifestations optimally, if not mandatorily, involves combined local and systemic treatment. However, it has been repeatedly reported that ENT manifestations, especially SGS, are frequently less, more slowly or only transiently responsive to systemic immunosuppressive therapy as compared with other manifestations such as alveolar haemorrhage or skin vasculitis [41, 45] and are often present at vasculitis relapse [46, 47]. Systemic treatment does not reverse cartilage erosion and tissue scarring of ENT lesions [48]. Thus, local treatment often becomes the fundamental and occasionally the sole treatment for some manifestations such as SGS [41]. In addition, courses of systemic antibiotics are often needed, particularly for GPA, to treat ENT suprainfections and/or bronchitis, alone or in combination with a brief increase in corticosteroid dose.
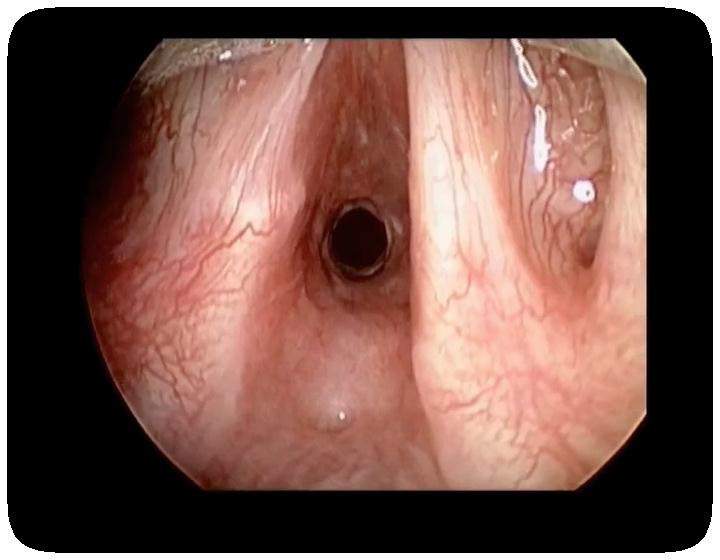
Figure 5
Direct laryngoscopy intraoperative view of subglottic stenosis in a patient with GPA (reprinted from Wolter NE, Ooi EH, Witterick IJ. Intralesional corticosteroid injection and dilatation provides effective management of subglottic stenosis in Wegener’s granulomatosis. Laryngoscope. 2010;120(12):2452–5. © 2010, with permission from John Wiley & Sons).
If EGPA does not affect the kidneys, heart or other major organ, systemic corticosteroids alone could be first-line therapy. Three-quarters to 90% of patients will achieve remission, but up to half will further experience disease exacerbation and require some additional immunosuppressant [49, 50]. Where the disease is more severe and/or generalised, an immunosuppressant, chiefly cyclophosphamide, is mandatory in combination with corticosteroids (see below, treatment of severe/generalised GPA, for further details on the therapeutic regimen, which also applies to patients with severe EGPA) [51].
For GPA, corticosteroids are mandatory and must be combined with an immunosuppressant to induce remission for all patients [52]. However, the strength of the combined immunosuppressant can be adjusted depending on the form of GPA (localised/limited/non-severe/early systemic versus severe/generalised/systemic). In a randomised controlled trial, cyclophophamide and methotrexate (15–25 mg per week) appeared to be equally effective in achieving remission, but concern regarding a high risk of relapse under methotrexate was suggested when patients stopped study treatment after 12 months [53]. Hence, if methotrexate is used with corticosteroids and controls the disease, it should be continued for several additional months, if not years. GPA patients carrying Staphylococcus aureus in nostrils are at increased risk of relapse and treatment with cotrimoxazole (trimethoprim 160 mg/sulfamethoxazole 800 mg twice a day) lowers the relapse rate by 40%, in combination with or after conventional immunosuppressive therapy, but the use of cotrimoxazole alone or in combination with corticosteroids is hardly ever sufficient [54]. In our opinion this latter strategy should clearly not be adopted otherwise than in referral centres for vasculitis and for a very few patients. In addition, cotrimoxazole, alone or combined with corticosteroids, was found to be inferior to methotrexate in maintaining remission in GPA [55].
For GPA patients with the severe/diffuse form, as well as those with severe EGPA, a strong immunosuppressive agent is needed in combination with corticosteroids. Cyclophosphamide is the classic first-line drug, given continuously and orally or intravenously, with the first 3 pulses administered every 2 weeks, then every 3 weeks until remission is achieved [56]. Subsequently patients require maintenance therapy with a less toxic drug, either azathioprine (2 mg/kg/d, orally) or methotrexate (0.3 mg/kg/week, orally or subcutaneously), for at least 2 years [57]. The optimal duration of this immunosuppressive maintenance regimen, as well as that of corticosteroid therapy, remains controversial.
Despite this conventional staged therapy, GPA relapse is frequent: 15% at 18 months post-diagnosis, 37% at 3 years, and 50% to 60% at 7 years [57]. Whether prolonging the duration of maintenance therapy (i.e., longer than 2 years) can lower the rate of relapse is under investigation (European Vasculitis Study Group REMAIN trial). Adjusting treatment (i.e. duration and/or intensity) based on individual patient characteristics could represent another option. Notably, the presence of ENT manifestations was found to be associated with a global low risk of death, for both GPA and EGPA, but associated with a higher risk of relapse in GPA, along with respiratory manifestations, antiPR3 ANCA positivity and a low serum creatinine level at diagnosis (<100 µmol/L) [47, 58–60]. Globally, GPA patients with ENT manifestations or lung nodules have more granulomatous disease and tend to have lower mortality but higher risk of relapse than those with lung haemorrhage or pauci-immune glomerulonephritis, their “vasculitic” counterparts [47].
Rituximab, a monoclonal antibody designed to deplete B cells, has recently been found to be a potential alternative to cyclophosphamide in inducing remission in adults with generalised forms of GPA and MPA, either newly diagnosed or relapsing. The double-blinded randomised RAVE trial aimed at comparing initial therapy with corticosteroids plus continuous oral cyclophosphamide (2 mg/kg/d until remission, then azathioprine for maintenance) or rituximab (375 mg/m2 on days 1, 8, 15 and 22, then placebo for maintenance) [61]. Almost 200 patients have been enrolled. Results at 6 months and 18 months suggested that rituximab was not inferior to staged cyclophosphamide-azathioprine regimen in obtaining remission and allowing corticosteroids to be stopped at 6 months (in several centres in North America corticosteroids are stopped at about month 6). The rate of adverse events between the 2 patient groups up to month 18 did not differ [62]. In view of the current cost of rituximab and the study’s failure to show any significant benefit in efficacy or short-term safety as compared with oral cyclophosphamide, rituximab use should probably be restricted to patients with refractory or relapsing disease or with contra-indications to cyclophosphamide or major concerns about its use. Response to rituximab in GPA patients with refractory and/or chronic granulomatous ENT lesions or orbital masses is variable and may take longer to achieve than for alveolar haemorrhage or renal disease [45, 63]. An important consideration is that the maintenance strategy following rituximab-based induction therapy is as yet undefined. Some centres use rituximab to re-treat only patients with a new disease flare; others give systematic re-infusions every 6 to 12 months, at variable doses, and yet others monitor B-cell levels to determine the need for re-infusion(s) [64].
Local treatment of URT manifestations of GPA or EGPA includes nasal saline rinses and topical corticosteroids. Rinses with mucolytics and other emollients, such as glycerine, can be tried in GPA patients with crusting rhinitis but to avoid the risk of lipoid or inhalation pneumonia must not be inhaled. Routine endoscopic nasal crust removal can sometimes benefit GPA patients. Surgical removal of nasal polyps in EGPA patients can also provide a certain relief, but of a transient nature in most cases because of early recurrence of the lesions or subsequent development of new ones. Nasal instillation, inhalation or irrigation with corticosteroids can be tried, especially for EGPA. Daily sino-nasal rinses and instillation or irrigation with antibiotic solutions have been used for a few GPA patients, with major benefit, but may foster the emergence of multi-resistant organisms. The application of antibiotic ointments (mupirocine, fucidine or bacitracine) to the anterior nares can be tried with GPA patients, particularly chronic carriers of S. aureus. Surgery should be proposed only after failure of conservative local management of chronic rhinosinusitis [9]. Functional endoscopic sinus surgery (FESS) for GPA represents a challenge to otorhinolaryngologists due to anatomical changes resulting from alteration of surgical landmarks by the disease process, ongoing or worsening mucosal inflammation, and post-operative issues with chronic crusting and unpredictable scarring [9]. Thus patient selection must be extremely cautious. If surgical management is pursued, FESS principles should be maintained. In particular, mucosal sparing techniques and preservation of retained structures are recommended [3, 9]. Meticulous post-operative care must be ensured, including daily saline irrigations, regular debridement of crusts and culture-directed antibiotic treatment [9].
Nasal reconstruction in GPA patients with saddle nose appears safe when performed with the disease in remission [65, 66]. The use of auricular, conchal or costal cartilage rather than irradiated material for grafting seems to be successful. A potential option, prior to such nasal reconstructive surgery, could be local injection of hyaluronic acid for transient nasal recontouring [67]. Nasal septum perforation repair is complicated due to the chronic relapsing nature of the disease, the frequently large size of the defect, and the poor surrounding sinus tissues which are unable to provide sustainable support for custom-fitted prostheses. With small-sized defects a silicone septal button can be considered [3].
Management of serous otitis media with effusion (OME) in GPA begins with thorough sino-nasal hygiene. Given that a significant proportion of OME is due to distal obstruction of the Eustachian tube, such findings should prompt endoscopic evaluation of the nasopharynx. Because according to some series otological manifestations of GPA seem to respond well to systemic therapy, initiation of surgical management should proceed with caution [3, 37]. Isolated episodes of serous OME may respond well to myringotomy tube placement when related to Eustachian tube dysfunction [37]. However, because chronic otitis media in GPA is caused by primary involvement of the middle ear and mastoid mucosa, tube insertion alone is insufficient and may result in aggravation of the otological lesions [37]. Therefore, tube insertion in the setting of GPA must be followed closely. Sensorineural hearing loss may be a sign of worsened disease and thus may require aggressive systemic treatment to address additional organ involvement. Once established, sensorineural hearing loss is usually insensitive to systemic therapy, although patients may benefit from hearing aids [68].
SGS can be life-threatening and should therefore be addressed early. Systemic treatment remains mandatory and is usually effective for generalised or severe GPA, but corticosteroids and conventional immunosuppressants usually have minimal effect on SGS. Corticosteroids can be transiently effective and remain indicated in patients with acute inflammatory exacerbations of SGS, but are unable to reverse scarring and fibrotic SGS. Data on cyclophosphamide or rituximab effects on SGS are scant [41, 48]. One study reported some efficacy of rituximab in 1 of only 2 patients and after a delay of 4 months post-infusion [69]; some responses were noted in a more recent study in all of the 11 patients who received this monoclonal antibody [70]. A potential limitation of these studies is the definition of responsiveness for SGS. Long-term follow-up, a minimum of 1 to 2 years, seems necessary to evaluate the response appropriately and determine whether rituximab (or another systemic) treatment can really limit the need for repeat dilatations.
Local management begins with clearance of secretions and meticulous care of the sino-nasal tract to avoid subsequent airway obstruction [41]. Crust formation may also be reduced with a humidified home environment. Given that many GPA patients with SGS also have laryngopharyngeal reflux, gastric acid suppression must be considered [71]. Surgical and/or endoscopy management may be necessary if respiratory symptoms progress. A number of procedures have been reported, including CO2 laser ablation [72], nitinol stenting [73] and intralesional corticosteroid injection and dilatation (ILCD) [33, 41, 71, 74]. Some authors have noted increased cicatricial scarring and poor post-operative outcomes in patients receiving CO2 laser treatment [33, 41, 71]. The most effective local procedure for treatment of SGS appears to be ILCD, with bougies or balloon dilators, followed by cold steel scar lysis with a sickle knife or endolaryngeal microscissors combined with in situ corticosteroid injection [33, 41, 75]. However, most GPA patients with SGS require several repeat procedures, in a recent study an average of 3 times at a mean interval of 11.5 months [74]. In these studies ILCD was indicated on the basis of symptoms regardless of disease severity and avoided tracheotomy [41, 74]. Spirometry parameters (forced expiratory volume in 1 second, forced vital capacity increase and peak expiratory flow rate) may be used to assess the response more objectively. Patients’ perceived improvement in breathing, along with a decrease in the rate of chest infections following the procedure(s), helps in evaluating the benefit of dilation(s). The utility of local applications of mitomycin-C, concomitant with dilations, is not well determined but is used on a regular basis by several groups. Reconstructive laryngotracheal surgery has been successful but is reserved for very severe cases or with failed endoscopy management and restricted to specialised centres [33, 76]. To ensure optimal success and minimal risk of complications, this surgical procedure must be performed only when the disease is in a quiescent phase, to avoid recurrence at the site, and in patients off, or taking only a minimal dose of, corticosteroids, to allow healing of the anastomosis.
| Table 1: Summary of the potential upper respiratory tract manifestations of the main systemic vasculitides. | ||
| Vasculitis | Main clinical characteristics | ENT/URT manifestations |
| ANCA-associated vasculitides (small-sized vessels) | ||
| Granulomatosis with polyangiitis (GPA; Wegener’s granulomatosis) | Three main target organs: ENT/URT, kidney (pauci-immune glomerulonephritis) and lungs (alveolar haemorrhage, parenchymal nodules) Two forms: localized/limited/non-severe/early systemic/non-systemic GPA and systemic/severe/generalized GPA Mainly associated with anti-PR3 C-ANCA | Serous otitis media: recurrent and/or chronic, possibly evolving to mastoiditis Middle-ear granulomatous lesions/tumours Conductive hearing loss Sensorineural hearing loss Rhinitis: almost constant, with nasal obstruction, crusting and epistaxis Sinusitis: sinus pain, frequently erosive (with bony erosion) and/or atrophic Nasal and/or sinus mucosa granulomatous inflammation, tumour and/or ulcers Anosmia/hyposmia Nasal cartilage erosion: saddle nose deformity, septum perforation, fistula Tongue ulcerations or infarctions Gingiva ulcerations or strawberry-like hypertrophy Sub-glottic stenosis |
| Churg–Strauss syndrome (eosinophilic granulomatosis with polyangiitis; EGPA) | Prior history of pseudo-allergic sinus polyposis and/or late-onset asthma Eosinophilia and eosinophilic tissue infiltration Anti-MPO P-ANCA in up to 1/3 of patients Cardiomyopathy: main factor of poor prognosis (more frequent in ANCA-negative patients) | Pseudo-allergic rhinitis, possibly with some crusting or epistaxis Nasal polyps Anosmia or hyposmia Sinusitis (non-erosive) |
| Microscopic polyangiitis (MPA) | Target organs: lungs (alveolar haemorrhage) and kidneys (pauci-immune glomerulonephritis) Mainly associated with antiMPO P-ANCA | Non-specific rhinitis or sinusitis, in up to 1/3 of patients, not erosive |
| Medium-sized-vessel vasculitides | ||
| Kawasaki disease | Vasculitis in children Fever, mucosal and skin manifestations, lymphadenopathies Risk of coronary artery aneurysms, especially if delayed diagnosis | Pharyngitis and cheilitis during initial phase |
| Polyarteritis nodosa | Can be related to hepatitis B virus infection Combination of several symptoms suggestive of vasculitis (fever, purpura, mononeuritis multiplex, abdominal pain, hypertension, orchitis etc.) Microaneurysms on abdomen and/or renal arteries on imaging (angiography or angio-CT scan) | Tongue necrosis (very rare) |
| Large-sized-vessel vasculitides | ||
| Giant cell arteritis | Adults aged >50 years Temporal tenderness and/or recent-onset headaches, jaw claudication Risk of (irreversible) ischaemic anterior optic neuritis | Tongue, lips or nasal septum infarction/necrosis: infrequent and consequence of local ischaemia |
| Takayasu arteritis | Young adult or teenager (aged <40 years), chiefly female Arm claudication, absence of a peripheral pulse on examination, asymmetrical blood pressure, subclavian or carotid bruits, hypertension (renal artery stenosis) | Nasal septum necrosis: rare and due to local perfusion ischaemia Carotidynia, spontaneous, at palpation and/or when swallowing: due to carotid vessel wall inflammation |
| ANCA = anti-neutrophil cytoplasm antibody (P- perinuclear or C- cytoplasmic labelling pattern on indirect immunofluorescence assay); ENT = ear, nose and throat; PR3 = proteinase 3; MPO = myeloperoxidase; URT = upper respiratory tract | ||
In other vasculitides, URT manifestations are rare, not specific and/or only transient (table 1). MPA is the third ANCA-associated vasculitis, with up to 90% of patients testing positive for ANCA, mainly anti-MPO P-ANCA, at diagnosis. Target organs are the lungs, with pulmonary capillaritis causing alveolar haemorrhage, and kidneys, with the same typical pauci-immune glomerulonephritis as in GPA or EGPA. ENT manifestations in MPA were reported in up to 30% of patients but are not specific and not erosive [6]. If erosive, the diagnosis of GPA may be considered, possibly with anti-MPO ANCA positivity.
In large-sized vessel vasculitides (giant cell arteritis and Takayasu arteritis), ENT lesions are infrequent and usually the result of local ischaemia around the neck and head arterial branches. A relatively typical manifestation of giant cell arteritis, though rare (less than 5% of patients), is tongue infarction and necrosis. Lips can also be involved. Nasal septum necrosis has been reported in a few patients with giant cell arteritis or Takayasu arteritis [77]. Patients with Takayasu arteritis may complain of antero-lateral neck pain, corresponding to carotidynia, when the disease is active and causing inflammation of the internal carotid(s).
Initial pharyngitis and cheilitis are signs of Kawasaki disease, the most frequent medium-sized vessel vasculitis in children, during the initial inflammatory phase, but are transient and benign. Rare cases of tongue necrosis have been reported in polyarteritis nodosa [78].
URT manifestations chiefly occur in GPA and EGPA and can present a real diagnostic challenge when initially isolated. Some ENT manifestations of vasculitis can inflict a severe burden on quality of life. Among all the possible URT manifestations, the one that gives most cause for concern, because it is potentially life-threatening and poorly responsive to available medications, is SGS. Despite appropriate systemic therapy for vasculitis, the ENT manifestations often tend to linger and are considered markers for increased risk of vasculitis relapse in general. Hopefully, systemic treatments under investigation or in development may be more effective. Until then, management must depend on close interaction and collaboration between otorhinolaryngologists, respirologists and internists or rheumatologists.
1 Falk RJ, Gross WL, Guillevin L, Hoffman GS, Jayne DR, Jennette JC, et al. Granulomatosis with polyangiitis (Wegener’s): an alternative name for Wegener’s granulomatosis. Arthritis Rheum. 2011;63(4):863–4.
2 Martinez Del Pero M, Sivasothy P. Vasculitis of the upper and lower airway. Best Pract Res Clin Rheumatol. 2009;23(3):403–17.
3 Erickson VR, Hwang PH. Wegener’s granulomatosis: current trends in diagnosis and management. Curr Opin Otolaryngol Head Neck Surg. 2007;15(3):170–6.
4 Hoffman GS, Kerr GS, Leavitt RY, Hallahan CW, Lebovics RS, Travis WD, et al. Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med. 1992;116(6):488–98.
5 Pagnoux C, Guillevin L. Churg-Strauss syndrome: evidence for disease subtypes? Curr Opin Rheumatol. 2010;22(1):21–8.
6 Guillevin L, Durand-Gasselin B, Cevallos R, Gayraud M, Lhote F, Callard P, et al. Microscopic polyangiitis: clinical and laboratory findings in eighty-five patients. Arthritis Rheum. 1999;42(3):421–30.
7 Pagnoux C, Guilpain P, Guillevin L. Churg-Strauss syndrome. Curr Opin Rheumatol. 2007;19(1):25–32.
8 Holle JU, Gross WL, Latza U, Nolle B, Ambrosch P, Heller M, et al. Improved outcome in 445 patients with Wegener’s granulomatosis in a German vasculitis center over four decades. Arthritis Rheum. 2011;63(1):257–66.
9 Cannady SB, Batra PS, Koening C, Lorenz RR, Citardi MJ, Langford C, et al. Sinonasal Wegener granulomatosis: a single-institution experience with 120 cases. Laryngoscope. 2009;119(4):757–61.
10 Lohrmann C, Uhl M, Warnatz K, Kotter E, Ghanem N, Langer M. Sinonasal computed tomography in patients with Wegener’s granulomatosis. J Comput Assist Tomogr. 2006;30(1):122–5.
11 Hoffman GS. Immunosuppressive therapy is always required for the treatment of limited Wegener’s granulomatosis. Sarcoidosis Vasc Diffuse Lung Dis. 1996;13(3):249–52.
12 Jennette JC, Falk RJ, Andrassy K, Bacon PA, Churg J, Gross WL, et al. Nomenclature of systemic vasculitides. Proposal of an international consensus conference. Arthritis Rheum. 1994;37(2):187–92.
13 Stone JH. Limited versus severe Wegener’s granulomatosis: baseline data on patients in the Wegener’s granulomatosis etanercept trial. Arthritis Rheum. 2003;48(8):2299–309.
14 Guillevin L, Lhote F, Brauner M, Casassus P. Antineutrophil cytoplasmic antibodies (ANCA) and abnormal angiograms in polyarteritis nodosa and Churg-Strauss syndrome: indications for the diagnosis of microscopic polyangiitis. Ann Méd Interne (Paris). 1995;146(8):548–50.
15 Sablé-Fourtassou R, Cohen P, Mahr A, Pagnoux C, Mouthon L, Jayne D, et al. Antineutrophil cytoplasmic antibodies and the Churg-Strauss syndrome. Ann Intern Med. 2005;143(9):632–8.
16 Sinico RA, Di Toma L, Maggiore U, Bottero P, Radice A, Tosoni C, et al. Prevalence and clinical significance of antineutrophil cytoplasmic antibodies in Churg-Strauss syndrome. Arthritis Rheum. 2005;52(9):2926–35.
17 Deng J, Ma-Krupa W, Gewirtz AT, Younge BR, Goronzy JJ, Weyand CM. Toll-like receptors 4 and 5 induce distinct types of vasculitis. Circ Res. 2009;104(4):488–95.
18 Summers SA, Steinmetz OM, Gan PY, Ooi JD, Odobasic D, Kitching AR, et al. Toll-like receptor 2 induces Th17 myeloperoxidase autoimmunity while Toll-like receptor 9 drives Th1 autoimmunity in murine vasculitis. Arthritis Rheum. 2011;63(4):1124–35.
19 Tadema H, Abdulahad WH, Stegeman CA, Kallenberg CG, Heeringa P. Increased expression of Toll-like receptors by monocytes and natural killer cells in ANCA-associated vasculitis. PLoS One. 2011;6(9):e24315.
20 Stegeman CA, Tervaert JW, Sluiter WJ, Manson WL, de Jong PE, Kallenberg CG. Association of chronic nasal carriage of Staphylococcus aureus and higher relapse rates in Wegener granulomatosis. Ann Intern Med. 1994;120(1):12–7.
21 Mahr AD, Neogi T, Merkel PA. Epidemiology of Wegener’s granulomatosis: Lessons from descriptive studies and analyses of genetic and environmental risk determinants. Clin Exp Rheumatol. 2006;24(2 Suppl 41):S82–91.
22 Pendergraft WF, 3rd, Pressler BM, Jennette JC, Falk RJ, Preston GA. Autoantigen complementarity: a new theory implicating complementary proteins as initiators of autoimmune disease. J Mol Med. 2005;83(1):12–25.
23 Voswinkel J, Muller A, Lamprecht P. Is PR3-ANCA Formation Initiated in Wegener’s Granulomatosis Lesions? Granulomas as Potential Lymphoid Tissue Maintaining Autoantibody Production. Ann N Y Acad Sci. 2005;1051:12–9.
24 Lane SE, Watts RA, Bentham G, Innes NJ, Scott DG. Are environmental factors important in primary systemic vasculitis? A case-control study. Arthritis Rheum. 2003;48(3):814–23.
25 Hogan SL, Satterly KK, Dooley MA, Nachman PH, Jennette JC, Falk RJ. Silica exposure in anti-neutrophil cytoplasmic autoantibody-associated glomerulonephritis and lupus nephritis. J Am Soc Nephrol. 2001;12(1):134–42.
26 Watts RA, Lane S, Scott DG. What is known about the epidemiology of the vasculitides? Best Pract Res Clin Rheumatol. 2005;19(2):191–207.
27 Mohammad AJ, Jacobsson LT, Westman KW, Sturfelt G, Segelmark M. Incidence and survival rates in Wegener’s granulomatosis, microscopic polyangiitis, Churg-Strauss syndrome and polyarteritis nodosa. Rheumatology (Oxford). 2009;48(12):1560–5.
28 Carrington CB, Liebow A. Limited forms of angiitis and granulomatosis of Wegener’s type. Am J Med. 1966;41(4):497–527.
29 Hoffman GS. Treatment of Wegener’s granulomatosis: time to change the standard of care? Arthritis Rheum. 1997;40(12):2099–104.
30 Hoffman GS, Langford CA. Are there different forms of life in the antineutrophil cytoplasmic antibody universe? Ann Intern Med. 2005;143(9):683–5.
31 Hoffman GS, Leavitt RY, Kerr GS, Fauci AS. The treatment of Wegener’s granulomatosis with glucocorticoids and methotrexate. Arthritis Rheum. 1992;35(11):1322–9.
32 Pagnoux C, Stubbe M, Lifermann F, Decaux O, Pavic M, Bérezné A, et al. Wegener’s granulomatosis strictly and persistently localized to one organ is rare: assessment of 16 patients from the French Vasculitis Study Group database. J Rheumatol. 2011;38(3):475–8.
33 Lebovics RS, Hoffman GS, Leavitt RY, Kerr GS, Travis WD, Kammerer W, et al. The management of subglottic stenosis in patients with Wegener’s granulomatosis. Laryngoscope. 1992;102(12 Pt 1):1341–5.
34 Rasmussen N. Management of the ear, nose, and throat manifestations of Wegener granulomatosis: an otorhinolaryngologist’s perspective. Curr Opin Rheumatol. 2001;13(1):3–11.
35 Fasunla JA, Hundt W, Lutz J, Forger F, Thurmel K, Steinbach S. Evaluation of smell and taste in patients with Wegener’s granulomatosis. Eur Arch Otorhinolaryngol. 2011.
36 Gomes GL, Halpern AS, Souza FH, Shinjo SK. Association between saddle nose deformity and retro-orbital mass in Wegener’s granulomatosis. Acta Reumatol Port. 2010;35(3):340–5.
37 Takagi D, Nakamaru Y, Maguchi S, Furuta Y, Fukuda S. Otologic manifestations of Wegener’s granulomatosis. Laryngoscope. 2002;112(9):1684–90.
38 Lourenco SV, Nico MM. Strawberry gingivitis: an isolated manifestation of Wegener’s granulomatosis? Acta Derm Venereol. 2006;86(1):90–1.
39 Holle JU, Gross WL, Holl-Ulrich K, Ambrosch P, Noelle B, Both M, et al. Prospective long-term follow-up of patients with localised Wegener’s granulomatosis: does it occur as persistent disease stage? Ann Rheum Dis. 2010;69(11):1934–9.
40 Peters JE, Salama AD, Ind PW. Wegener’s granulomatosis presenting as acute systemic vasculitis following 20 years of limited tracheobronchial disease. J Laryngol Otol. 2009;123(12):1375–7.
41 Langford CA, Sneller MC, Hallahan CW, Hoffman GS, Kammerer WA, Talar-Williams C, et al. Clinical features and therapeutic management of subglottic stenosis in patients with Wegener’s granulomatosis. Arthritis Rheum. 1996;39(10):1754–60.
42 Bacciu A, Bacciu S, Mercante G, Ingegnoli F, Grasselli C, Vaglio A, et al. Ear, nose and throat manifestations of Churg-Strauss syndrome. Acta Otolaryngol. 2006;126(5):503–9.
43 Devaney KO, Travis WD, Hoffman G, Leavitt R, Lebovics R, Fauci AS. Interpretation of head and neck biopsies in Wegener’s granulomatosis. A pathologic study of 126 biopsies in 70 patients. Am J Surg Pathol. 1990;14(6):555–64.
44 Del Buono EA, Flint A. Diagnostic usefulness of nasal biopsy in Wegener’s granulomatosis. Hum Pathol. 1991;22(2):107–10.
45 Holle JU, Dubrau C, Herlyn K, Heller M, Ambrosch P, Noelle B, et al. Rituximab for refractory granulomatosis with polyangiitis (Wegener’s granulomatosis): comparison of efficacy in granulomatous versus vasculitic manifestations. Ann Rheum Dis. 2011.
46 Koldingsnes W, Nossent JC. Baseline features and initial treatment as predictors of remission and relapse in Wegener’s granulomatosis. J Rheumatol. 2003;30(1):80–8.
47 Pierrot-Deseilligny Despujol C, Pouchot J, Pagnoux C, Coste J, Guillevin L. Predictors at diagnosis of a first Wegener’s granulomatosis relapse after obtaining complete remission. Rheumatology (Oxford). 2010;49(11):2181–90.
48 Langford CA. Update on the treatment of granulomatosis with polyangiitis (Wegener’s). Curr Treat Options Cardiovasc Med. 2012. [Epub ahead of print]
49 Ribi C, Cohen P, Pagnoux C, Mahr A, Arène JP, Lauque D, et al. Treatment of Churg-Strauss syndrome without poor-prognosis factors: A multicenter, prospective, randomized, open-label study of seventy-two patients. Arthritis Rheum. 2008;58(2):586–94.
50 Ribi C, Cohen P, Pagnoux C, Mahr A, Arene JP, Puechal X, et al. Treatment of polyarteritis nodosa and microscopic polyangiitis without poor-prognosis factors: A prospective randomized study of one hundred twenty-four patients. Arthritis Rheum. 2010;62(4):1186–97.
51 Guillevin L, Lhote F, Gayraud M, Cohen P, Jarrousse B, Lortholary O, et al. Prognostic factors in polyarteritis nodosa and Churg-Strauss syndrome. A prospective study in 342 patients. Medicine (Baltimore). 1996;75(1):17–28.
52 Mukhtyar C, Guillevin L, Cid MC, Dasgupta B, de Groot K, Gross W, et al. EULAR recommendations for the management of primary small and medium vessel vasculitis. Ann Rheum Dis. 2009;68(3):310–7.
53 de Groot K, Rasmussen N, Bacon PA, Tervaert JW, Feighery C, Gregorini G, et al. Randomized trial of cyclophosphamide versus methotrexate for induction of remission in early systemic antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum. 2005;52(8):2461–9.
54 Reinhold-Keller E, De Groot K, Rudert H, Nolle B, Heller M, Gross WL. Response to trimethoprim/sulfamethoxazole in Wegener’s granulomatosis depends on the phase of disease. QJM. 1996;89(1):15–23.
55 de Groot K, Reinhold-Keller E, Tatsis E, Paulsen J, Heller M, Nolle B, et al. Therapy for the maintenance of remission in sixty-five patients with generalized Wegener’s granulomatosis. Methotrexate versus trimethoprim/sulfamethoxazole. Arthritis Rheum. 1996;39(12):2052–61.
56 de Groot K, Harper L, Jayne DR, Flores Suarez LF, Gregorini G, Gross WL et al. Pulse versus daily oral cyclophosphamide for induction of remission in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized trial. Ann Intern Med. 2009;150(10):670–80.
57 Pagnoux C, Mahr A, Hamidou MA, Boffa JJ, Ruivard M, Ducroix JP, et al. Azathioprine or methotrexate maintenance for ANCA-associated vasculitis. N Engl J Med. 2008;359(26):2790–803.
58 Walsh M, Flossmann O, Berden A, Westman K, Hoglund P, Stegeman C, et al. Risk factors for relapse of ANCA associated vasculitis. Arthritis Rheum. 2012;64(2):542–8.
59 Pagnoux C, Hogan SL, Chin H, Jennette JC, Falk RJ, Guillevin L, et al. Predictors of treatment resistance and relapse in antineutrophil cytoplasmic antibody-associated small-vessel vasculitis: Comparison of two independent cohorts. Arthritis Rheum. 2008;58(9):2908–18.
60 Guillevin L, Pagnoux C, Seror R, Mahr A, Mouthon L, Le Toumelin P. The Five-Factor Score revisited: assessment of prognoses of systemic necrotizing vasculitides based on the French Vasculitis Study Group (FVSG) cohort. Medicine (Baltimore). 2011;90(1):19–27.
61 Stone JH, Merkel PA, Spiera R, Seo P, Langford CA, Hoffman GS, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med. 2010;363(3):221–32.
62 Stone JH, Merkel PA, Seo P, Spiera R, Langford CA, Hoffman GS, et al. Extended follow-up of treatment with rituximab versus cyclophosphamide for remission-induction of ANCA-associated vasculitis: Which subsets are at greatest risk for flare? Arthritis Rheum. 2011;63(10 Suppl.):S946–7.
63 Aouba A, Pagnoux C, Bienvenu B, Mahr A, Guillevin L. Analysis of Wegener’s granulomatosis responses to rituximab: current evidence and therapeutic prospects. Clin Rev Allergy Immunol. 2008;34(1):65–73.
64 Cartin-Ceba R, Fervenza FC, Specks U. Treatment of antineutrophil cytoplasmic antibody-associated vasculitis with rituximab. Curr Opin Rheumatol. 2012;24(1):15–23.
65 Vogt PM, Gohritz A, Haubitz M, Steiert A. Reconstruction of nasal deformity in Wegener’s granulomatosis: contraindication or benefit? Aesthetic Plast Surg. 2011;35(2):156–61.
66 Congdon D, Sherris DA, Specks U, McDonald T. Long-term follow-up of repair of external nasal deformities in patients with Wegener’s granulomatosis. Laryngoscope. 2002;112(4):731–7.
67 Bennett HS, Reilly PG. Restylane – a temporary alternative for saddle nose deformity in nasal Wegener’s granulomatosis--how we do it. Br J Oral Maxillofac Surg. 2011;49(4):e3–5.
68 Bakthavachalam S, Driver MS, Cox C, Spiegel JH, Grundfast KM, Merkel PA. Hearing loss in Wegener’s granulomatosis. Otol Neurotol. 2004;25(5):833–7.
69 Aries PM, Hellmich B, Voswinkel J, Both M, Nolle B, Holl-Ulrich K, et al. Lack of efficacy of rituximab in Wegener’s granulomatosis with refractory granulomatous manifestations. Ann Rheum Dis. 2006;65(7):853–8.
70 Martinez Del Pero M, Chaudhry A, Jones RB, Sivasothy P, Jani P, Jayne D. B-cell depletion with rituximab for refractory head and neck Wegener’s granulomatosis: a cohort study. Clin Otolaryngol. 2009;34(4):328–35.
71 Gluth MB, Shinners PA, Kasperbauer JL. Subglottic stenosis associated with Wegener’s granulomatosis. Laryngoscope. 2003;113(8):1304–7.
72 Shvero J, Shitrit D, Koren R, Shalomi D, Kramer MR. Endoscopic laser surgery for subglottic stenosis in Wegener’s granulomatosis. Yonsei Med J. 2007;48(5):748–53.
73 Watters K, Russell J. Subglottic stenosis in Wegener’s granulomatosis and the nitinol stent. Laryngoscope. 2003;113(12):2222–4.
74 Wolter NE, Ooi EH, Witterick IJ. Intralesional corticosteroid injection and dilatation provides effective management of subglottic stenosis in Wegener’s granulomatosis. Laryngoscope. 2010;120(12):2452–5.
75 Eliachar I, Chan J, Akst L. New approaches to the management of subglottic stenosis in Wegener’s granulomatosis. Cleve Clin J Med. 2002;69(Suppl 2):SII-149–51.
76 Herridge MS, Pearson FG, Downey GP. Subglottic stenosis complicating Wegener’s granulomatosis: surgical repair as a viable treatment option. J Thorac Cardiovasc Surg. 1996;111(5):961–6.
77 Akar S, Dogan E, Goktay Y, Can G, Akkoc N, Sarioglu S, et al. Nasal septal perforation in a patient with Takayasu’s arteritis; a rare association. Intern Med. 2009;48(17):1551–4.
78 Buonuomo PS, El Hachem M, Callea F, Bracaglia C, Diociaiuti A, Pardeo M, et al. Necrosis of the tongue as first symptom of Polyarteritis Nodosa (PAN): unusual presentation of a rare disease in children. Rheumatol Int. 2010. [Epub ahead of print]
Funding / potential competing interests: No financial support relevant to this article needs to be reported. Over the past 5 years, Dr. Pagnoux received lecture fees from Hoffmann-La Roche (<USD10,000) and LFB (Laboratoire français du Fractionnement et des Biotechnologies, <USD10,000) and consulting fees from Schering-Plough (<USD10,000).