
DOI: https://doi.org/10.4414/smw.2012.13519
The term “windswept deformity” describes the appearance of an abnormal valgus deformity in one knee in association with varus in the other. It is commonly seen in young children in certain parts of Africa, and it develops secondary to metabolic bone diseases. Metabolic bone diseases frequently result in skeletal deformities, especially in the legs, due to physeal disturbances or defective mineralisation in children before puberty. Among these disorders, hypophosphataemic rickets, hypophosphatasia, and renal osteodystrophy are the most frequent [1–6].
Schwartz-Jampel syndrome (SJS) is a term now applied to 2 different autosomal recessive inherited conditions, sometimes termed SJS type I and SJS type II. Both are very rare. SJS type I has 2 recognised subtypes, IA and IB, which are similar except that type IB manifests earlier and with greater severity. The most commonly recognised and described type is IA, which exhibits muscle stiffness, mild (and largely non-progressive) muscle weakness, and a number of minor morphological abnormalities. In affected patients, problems with motor development frequently become evident during the first year of life. Usually, the characteristic dysmorphic features lead to an early diagnosis, no later than 3 years of age. Myotonia results in a fixed facial expression with pursed lips and narrowed palpebral fissures.
Types IB and type II are now known to be a separate disease more commonly referred to as Stuve-Wiedemann syndrome [7–10]. In the reported patient, a combination of orthopaedic interventions were carried out in order to correct the windswept leg deformity.
A 13-year-old boy who presented with the full diagnostic criteria of Schwartz-Jampel syndrome was referred to our department to manage his recurrent and unstable genu valgum on the left side and genu varum on the right side (fig. 1). Dysmorphic facial features, bilateral congenital cataracts, blepharophimosis with stiff facial muscles and an expressionless dull face were notable. He presented with trismus (spasm around the mouth making it difficult to open on command, and the mouth was involuntary held in a fixed half-smile (fig. 2). He had been previously treated in other orthopaedic centres and the outcome was unpleasant. He was born at full term as a product of an uncomplicated gestation to a healthy first cousin. At birth he manifested severe muscle stiffness, and joint deformities at the hips, knees and ankles effectively causing the development of excessive genu valgum. In his first year of life, phenotypic characterisation and genetic testing confirmed the diagnosis. His blood differential and chemistry were normal, with the exception of creatine kinase, which was elevated at 720 I/U. The electromyogram showed occasional myotonic discharges. The muscle biopsy was non-contributory. The clinical diagnosis was confirmed by means of molecular analysis and it showed compound heterozygosity with mutations in the two Perlecan alleles. Bilateral corneal opacities were dealt with by means of corneal transplantation performed at the age of 6 years. As the patient was treated in a different orthopaedic institute in which he had undergone resection of the femoral head of the left side, the proximal femoral osteotomy was performed as a pelvic support osteotomy (valgus osteotomy). A distal femoral osteotomy and a proximal tibial osteotomy were performed as well. Osteosynthesis was provided by the use of an external fixation system for the femur and the tibia (Taylor Spatial Frame™, Smith & Nephew company, Memphis, US) to perform a gradual correction of the valgus deformity which was 70° (fig. 3, 4). Unfortunately, during the corrective process, weakness of the peroneal muscles and the long extensors were the outcome. Corrective operations at our institution started for the first time at the age of 13 years on the left side. After bony consolidation of the osteotomies and after having achieved corrections of the mal-alignment, the Taylor Spatial Frame™ was removed. To prevent a fracture, a TEN was inserted into the medullary cavity of the tibia.
Six months later and because of the severe varus deformity of the right femur (30°) and anti-curvation, corrective osteotomies were performed at the right distal femur and proximal tibia, and a Taylor Spatial Frame™ was mounted thereafter (fig. 5). Three months later after full correction and full bony consolidation, the device was removed and an intramedullary stabilisation using TEN nails was applied (fig. 6). Simultaneously, both knees showed severe instability of the medial collateral ligament, and therefore the patient was treated with above-knee-orthoses.
Angular deformities of the knee resulting from idiopathic, congenital or acquired causes are commonly encountered in paediatric orthopaedics. A noticeable number of parents bring their children to orthopaedic clinics because of angular deformities at the knees. The deformity may be either unilateral or bilateral (often termed bow legs or knock knees), or combining two components as seen in “windswept deformity”. Salawu published a 70% rickets rate among the 103 cases examined. Fulford and Brown applied the term “windswept deformity” in a different context to a postural deformity acquired by spastic infants during the first few weeks of life. Blount described a condition of tibia vara in children and called it osteochondrosis deformans tibiae, and Golding reported about an extensive survey of this condition in West Indian children. Both authors recognised a more common infantile form arising in the first, second or third year of life and a rarer, usually less severe, adolescent type. The deformity could be unilateral or bilateral and no certain aetiological factor was identified. Therefore, it is imperative, when evaluating a patient with angular deformity of the knee, that the orthopaedic surgeon must keep several principles in mind. The basic principle is to establish a correct diagnosis [1–6, 11, 12].
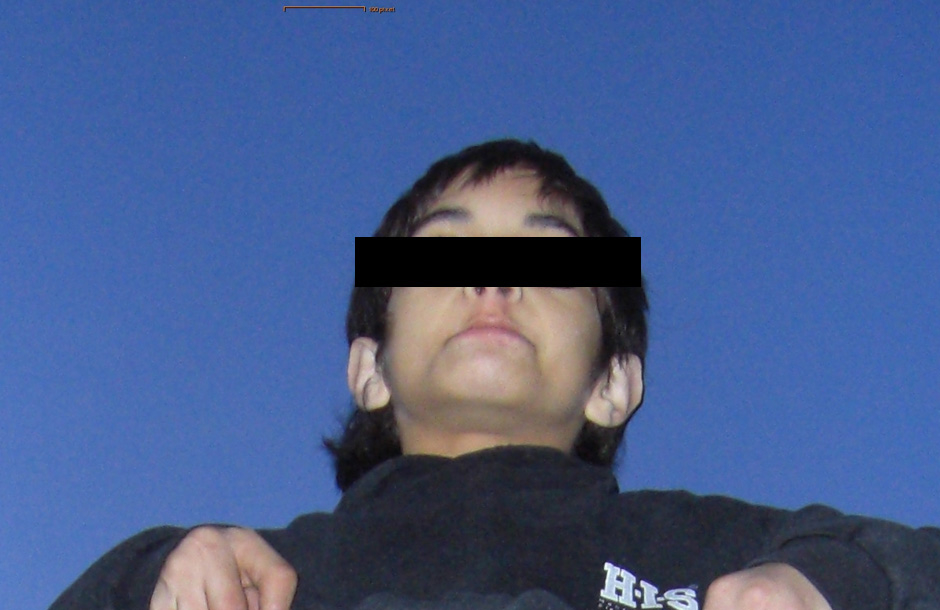
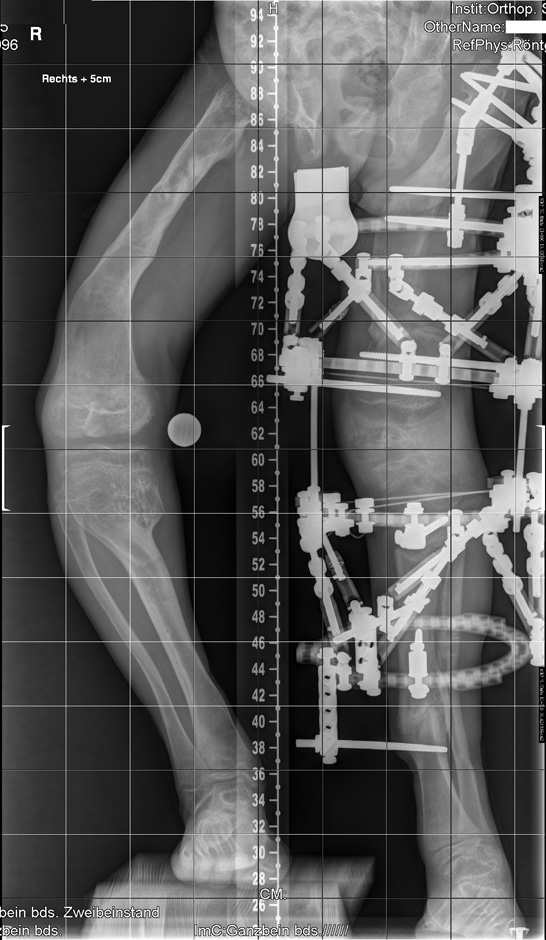
Figure 1
AP-view weight bearing X-ray of the patient before starting correction showed, at the pelvis and the femora, severe dysplasia of the capital femoral epiphyses, the acetabulae and the hypoplastic pelvis associated with bowing of the upper femoral shaft and Erlenmeyer flask appearance of the lower femora. Note the windswept deformity which presents abnormal valgus deformity in one knee in association with varus deformity in the other. The inferior femoral and superior tibial epiphyses look enlarged and severely distorted.
Figure 2
Dysmorphic facial features, bilateral congenital cataract, blepharophimosis with stiff facial muscles and an expressionless dull face are notable. Trismus is also present (spasm around the mouth making it difficult to open on command), and the mouth is involuntary held in a fixed half-smile. (The patient's father has given written informed consent for publication.)
Figure 3
After performing a proximal and distal femoral osteotomy and a proximal tibial osteotomy on the left side, a Taylor Spatial Frame™ was mounted.
The management of patients with multi-apical bony deformities with established metabolic bone diseases is complex. The deformities are either discrete and angular, or long-bowing (multi-apical) deformities. Angular deformities originate from or adjacent to the growth plate and often a single osteotomy is required to correct the deformity. Multi-apical deformities usually result from bowing of the entire long bone. Frequently, more than one osteotomy is needed to correct the deformity in order to produce a straight bone and avoid secondary iatrogenic deformities [2].
Osteotomy and stabilisation with Ilizarov type external fixators allows gradual, controlled correction of a deformity with the advantages of high union and low infection rates because of the low energy involved in osteotomy and minimal intra-osseous fixation. The Ilizarov technique allows post-operative adjustment and prevents inequality of limb length [6].
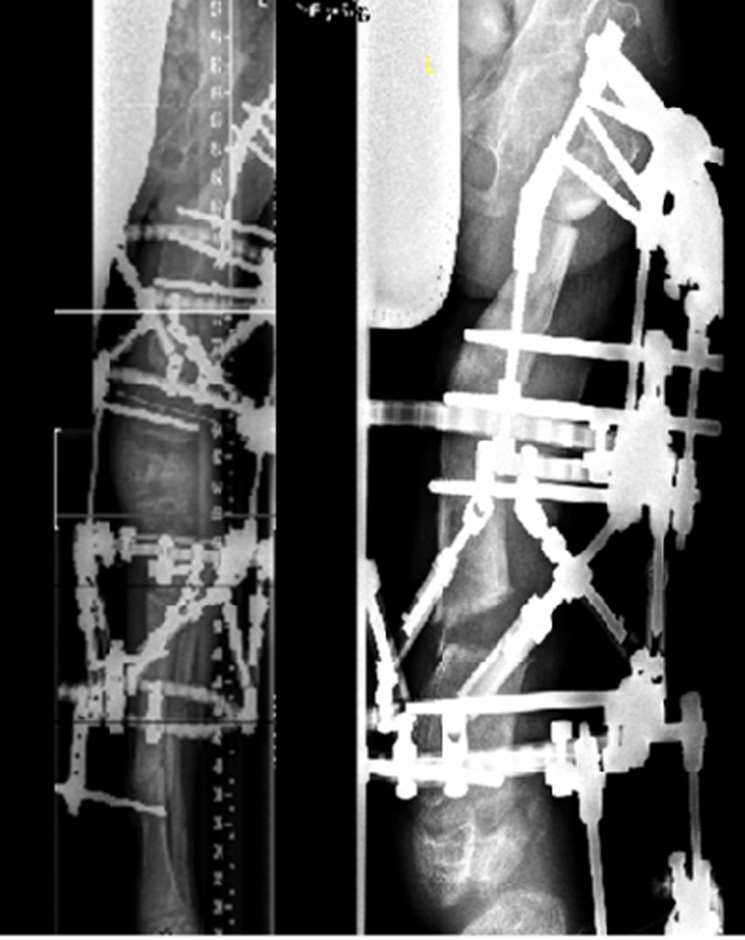
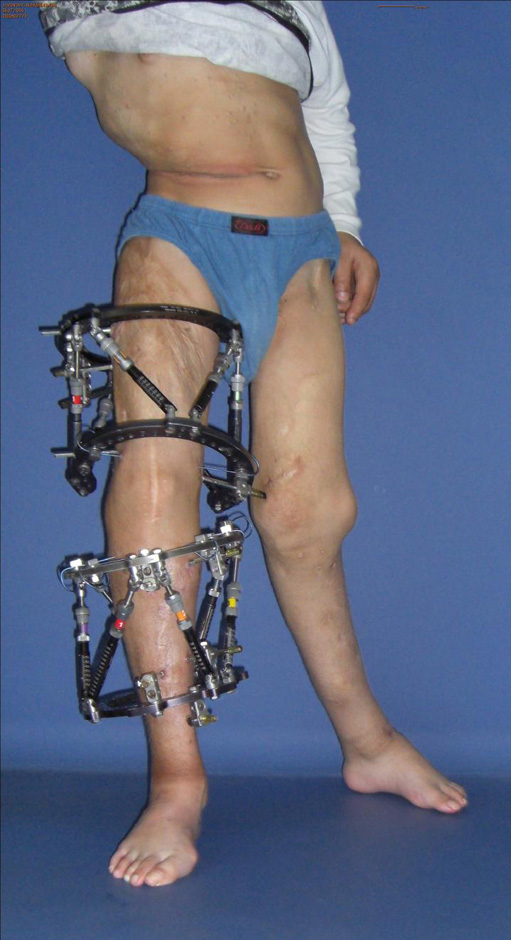
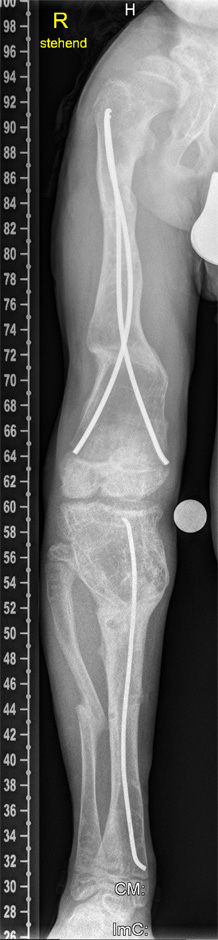
Figure 4
Three months later, the deformity of the left side was fully corrected by gradual distraction.
Figure 5
After distal femoral and proximal tibial osteotomy, the severe varus deformity was corrected by gradual distraction using the Taylor Spatial Frame™.
Figure 6
After consolidation and removal of the frame, a prophylactic nailing of the right femur and tibia was performed to obtain the correction.
The Taylor Spatial Frame™ uses the slow correction principles of the Ilizarov system but adds a six-axis deformity analysis incorporated within a computer programme. Chronic deformity applies to congenital deformity, malunions, and stiff malunions that can be measured fairly accurately by radiographs. Based on these radiographs and clinical examination, a spatial frame can be adjusted from its natural or home position (at which all struts are of equal length) to a deformed position that exactly matches the skeletal deformity. The spatial frame is then fixed to the skeleton. As the frame is returned to its neutral or home position, the fragments are restored to their anatomic positions. This process is called chronic deformity correction [13–15].
Oscar Schwartz and Robert Jampel jointly described a rare autosomal recessive disorder in a pair of siblings. The children had short stature, myotonia with paucity of facial expression, blepharophimosis, pectus carinatum and contractures. This disorder was also designated osteo-chondro-muscular dystrophy or chondrodystrophic myotonia, and it was initially thought to be neurogenic in aetiology. To date, there have been several reports in Middle Eastern families and South African kindreds, the prevalence of which may be accounted for by parental consanguinity in these regions. The characteristic features include myotonia with a fixed or frozen facial expression, pursed lips and a small mouth. The ocular manifestations are blepharophimosis, myopia and medial displacement of outer canthi. Patients have a waddling gait and frequently adopt a crouched stance because of joint stiffness. The skeletal findings are short stature, pectus carinatum, kyphoscoliosis, platyspondyly with coronal clefts in the vertebrae, metaphyseal and epiphyseal dysplasia, and joint contractures. Respiratory and feeding difficulties may occur shortly after birth. Mild mental retardation, which may be intrinsic to the syndrome or a result of the severity of physical limitations, has been said to be present in about 25% of cases. Muscle hypertrophy is evident in most patients. Malignant hyperthermia during anaesthesia is a potentially lethal hazard. In the prenatal period, polyhydramnios and absence of a stomach bubble, both indicative of a swallowing disorder, may be notable [7–10].
Nicole et al. [16] demonstrated missense and splicing mutations in the HSPG2 gene encoding perlecan in this type. This is a heparan sulphate proteoglycan highly expressed in basement membranes and cartilage. Superti-Furga et al. and Cormier-Daire et al. [17, 18] also suggest that Stuve-Wiedemann syndrome and Schwartz-Jampel syndrome type 2 are allelic conditions.
The Taylor Spatial Frame™ (TSF) is a known tool for external ring fixation system developed for correction of axial, sagittal, planar and rotational deformities of the extremities [19]. A specialised feature of the TSF is its virtual hinge, which allows for the simultaneous gradual correction of multi-planar deformities and limb lengthening through one osteotomy site. The power of the spatial frame lies in its precise control over the final limb length and alignment and its ability to correct a residual deformity. The stability of this multi-planar circular fixator permits early weight-bearing and provides an ideal environment for both new-bone formation and soft–tissue healing [20, 21]. Finally, we wish to stress that the poor bone quality in patients with skeletal dysplasias makes optimal correction and re-alignment extremely difficult.
1 Smyth EHJ. Windswept deformity. J Bone Joint Surg. (Br) 1980;62-B:166–7.
2 Mankin HJ. Rickets, osteomalacia and renal osteodystrophy: an update. Orthop Clin North Am. 1990;21:81–96.
3 Fulford GE, Brown JK. Position as a cause of deformity in children with cerebral palsy. Dev Med Child Neurol. 1976;18:305–14.
4 Golding JSR. Tibia vara. J Bone Joint Surg. (Br). 1962;44-B:216.
5 Blount WP. Tibia vara. J Bone Joint Surg. 1937;19:1–29.
6 Stanitski DF. Treatment of deformity secondary to metabolic bone disease with Ilizarov technique. Clin Orthop. 1994;301:38–41.
7 Schwartz O, Jampel RS. Congenital blepharophimosis associated with a unique generalized myopathy. Arch Ophthalmol. 1962;68:52–7.
8 Di Rocco M, Stella G, Bruno C, et al. Long-term survival in Stuve-Wiedemann syndrome: a neuro-myo-skeletal disorder with manifestations of dysautonomia. Am J Med Genet A. 2003;118(4):362–8.
9 Stum M, Davoine CS, Vicart S, et al. Spectrum of HSPG2 (Perlecan) mutations in patients with Schwartz-Jampel syndrome. Hum Mutat. 2006;27(11):1082–91.
10 Rodgers KD, Sasaki T, Aszodi A, Jacenko O. Reduced perlecan in mice results in chondrodysplasia resembling Schwartz-Jampel syndrome. Hum Mol Genet. 2007;16(5):515–28.
11 Salawu SAI. Knock knee and bow legs in Zaria. Orient Journal of Medicine. 1992;4:69–72.
12 Salenius P, Vankka E. The development of the tibio-femoral angle in children. J Bone Joint Surg. 1975;57-A:259–261.
13 Manner HM, Huebl M, Radler C, Ganger R, Petje G, Grill F. Accuracy of complex lower-limb deformity correction with external fixation: a comparison of the Taylor Spatial Frame with the Ilizarov Ringfixator. J Child Orthop. 2007;1:55–61.
14 Naqui SZ, Thiryayi W, Foster A. Tselentakis G, Evans M, Day JB. Correction of simple and complex pediatric deformities using the Taylor-Spatial Frame. J Pediatr Orthop. 2008;28(6):640–7.
15 Rozbruch SR, Fragomen AT, Ilizarov S. Correction of tibial deformity with use of the Ilizarov-Taylor spatial frame. J Bone Joint Surg Am. 2006;88(Suppl 4):156–74.
16 Nicole S, Davoine CS, Topaloglu H, et al. Perlecan, the major proteoglycan of basement membranes, is altered in patients with Schwartz-Jampel syndrome (chondrodystrophic myotonia). Nat Genet. 2000;26(4):480–3.
17 Superti-Furga A, Tenconi R, Clementi M, Eich G, Steinmann B, Boltshauser E, Giedion A. Schwartz-Jampel syndrome type 2 and Stuve-Wiedemann syndrome: a case for “lumping”. Am J Med Genet. 1998;78:150–4.
18 Cormier-Daire V, Superti-Furga A, Munnich A, Lyonnet S, Rustin P, Delezoide AL, et al. Clinical homogeneity of the Stuve-Wiedemann syndrome and overlap with the Schwartz-Jampel syndrome type 2. Am J Med Genet. 1998;78:146–9.
19 Manner HM, Huebl M, Radler C, Ganger R, Petje G, Grill F. Accuracy of complex lower-limb deformity correction with external fixation: a comparison of the Taylor Spatial Frame with the Ilizarov Ringfixator. J Child Orthop. 2007;1:55–61.
20 Naqui SZ, Thiryayi W, Foster A, Tselentakis G, Evans M, Day JB. Correction of simple and complex pediatric deformities using the Taylor-Spatial Frame. J Pediatr Orthop. 2008;28(6):640–7.
21 Rozbruch SR, Fragomen AT, Ilizarov S. Correction of tibial deformity with use of the Ilizarov-Taylor spatial frame. J Bone Joint Surg Am. 2006;88(Suppl 4):156–74.
Funding / potential competing interests: No financial support and no other potential conflict of interest relevant to this article were reported.
1 Written informed consent allowing publication of this case has been obtained from the patient's father.