Figure 1
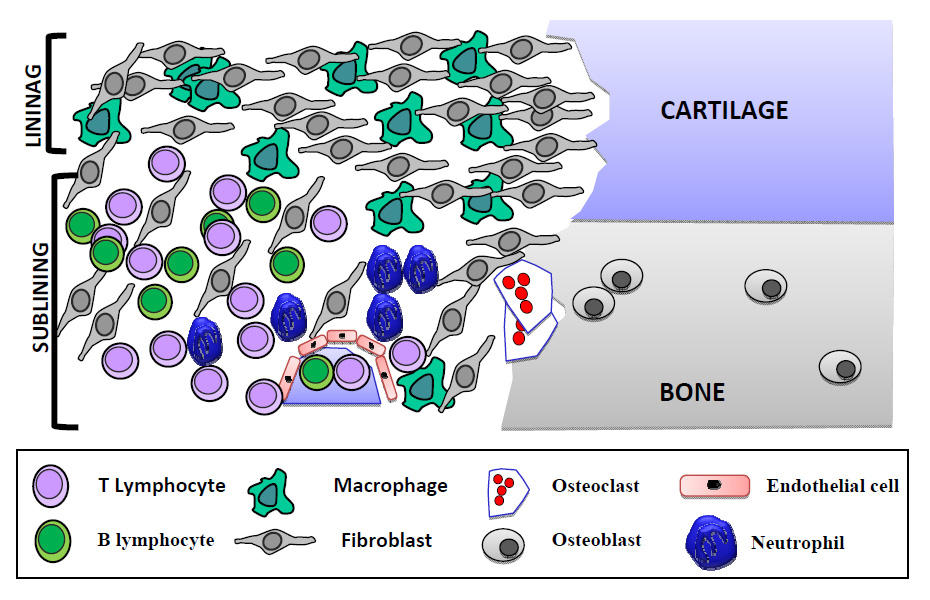
Representation of the different cell types in the rheumatoid synovium.
DOI: https://doi.org/10.4414/smw.2012.13529
RA is an autoimmune disease, characterised by the development of innate and adaptive immune responses and, in a high percentage of patients, the presence of autoantibodies (rheumatoid factor and anti-cyclic citrullinated peptide antibodies) which may be detected in blood many years before disease onset and whose pathogenic role remains unclear [6]. Additionally, genetic factors such as most notably the presence of the shared epitope (a specific group of HLA-DR4 alleles) and single nucleotide polymorphisms in the PTPN22 gene are known to play a part in disease susceptibility and severity [8]. A number of environmental factors have also been described. The association of smoking with disease development in individuals who carry the shared epitope and worse disease outcome is well established [9]. Local factors such as hypoxia, which is thought to be important in immune signalling and angiogenesis, are also relevant. However, a unified theory on how these factors interact together is still elusive (for a review on this topic see reference [10]).
Figure 1
Representation of the different cell types in the rheumatoid synovium.
At a local joint level, RA is characterised by a radical change of the two compartments of the synovium. The lining layer of the tissue situated adjacent to the synovial fluid compartment undergoes dramatic hyperplasia, sometimes reaching 10–15 cells in depth with expansion of both macrophage and fibroblast populations. At the articular borders the thickened synovial lining layer may become a mass of “pannus” tissue (rich in fibroblasts and osteoclasts) that invades the adjacent articular cartilage and subchondral bone. The sublining layer also undergoes expansion, with infiltrates of inflammatory cells including macrophages, mast cells, T cells, B cells and plasma and dendritic cells. T and B lineage cells may remain in diffuse infiltrates, or may coalesce into aggregates of cells varying from simple perivascular “cuffs” a few cells in diameter to structures resembling B-cell follicles in up to 20% of samples [11]. This increased activity is supported by further extracellular matrix (ECM) production and neoangiogenesis, although the inflamed synovium remains in a state of relative hypoxia [12].
The interactions between these different cell types are complex and mediated by numerous cytokine networks. The description of the relative contribution of each of these cell types to disease pathogenesis is beyond the remit of this paper but a schematic representation of the synovium’s cellularity in RA has been depicted in figure 1.
Over the last decade, researchers have demonstrated that in addition to their well-known structural role in “landscaping” the microenvironment, synovial fibroblasts are implicated in the pathogenesis of RA at two fundamental levels.
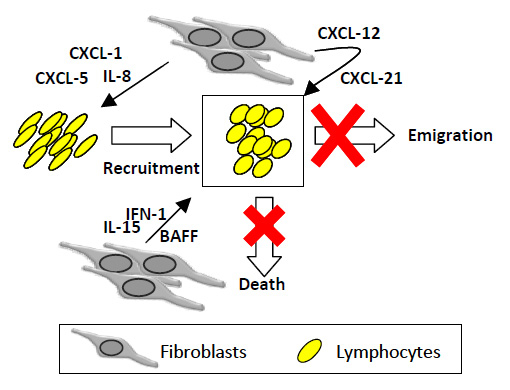
Figure 2
Fibroblasts orchestrate chronic inflammation in RA. The maintenance of persistent leukocyte infiltrates results from a distorted homeostatic balance between leukocyte recruitment, proliferation, emigration and death. Activated RASFs produce inflammatory chemokines (IL-8, CXCL5, CXCL1) implicated in leukocyte recruitment to diseased synovium, survival factors (type I interferon, IL-15, BAFF) and constitutive chemokines (e.g. CXCL12, CCL21). The net result is the chronic accumulation, survival and retention of leukocytes at sites of disease.
Firstly, activated RA synovial fibroblasts play crucial roles in determining the site at which inflammation occurs, and in the subsequent maintenance of persistent inflammation in the joint microenvironment, as reflected in hyperplasia of resident stromal cells and the organisation of T and B lymphocyte infiltration discussed above [11]. Much work has been devoted to the description of this microenvironment and of the complex haematopoietic-stromal cell interactions that determine its stability [3–5]. We have proposed that the maintenance of the persistent leukocyte infiltrate results from a distorted homeostatic balance between leukocyte recruitment, proliferation, emigration and death [3], and that synovial fibroblasts are key determinants of this imbalance. In the rheumatoid synovium, different subpopulations of stromal cells exist. Stromal cells of the RA synovium differ from those of the normal synovium. Additionally synovial fibroblasts from the lining layer differ in biological and morphological characteristics to those of the sublining layer. The so-called activated synovial fibroblasts found in RA tissue (RASFs) are the key stabilisers of the abnormal synovial microenvironment. In a series of experiments, our group and others have demonstrated that activated RASFs produce inflammatory chemokines (e.g., IL-8, CCL5, CXCL1) [13, 14] that actively recruit leukocytes such as neutrophils and B and T lymphocytes to the diseased synovial tissue. By contrast RASFs also inappropriately secrete so-called constitutive chemokines (e.g., CXCL12, CXCL13, CCL21) that are normally restricted to lymphoid tissues in order to coordinate the recirculation of lymphocytes [3, 15–18]. In this way, infiltrating leukocytes are prevented from leaving the synovial tissue. RASFs also produce survival factors (e.g., type I interferon, IL-15, BAFF) that prevent leukocyte subpopulations from dying by apoptosis as would normally occur during the resolution phase of an inflammatory response [19–21]. Thus fibroblast activation results in the accumulation, survival and retention of leukocytes at sites of disease, mimicking the microenvironment seen in lymphoid tissues, and thereby preventing the resolution of chronic inflammation (fig. 2).
Secondly, RASFs are key mediators of cartilage and bone destruction. They act directly on cartilage via secretion of matrix metalloproteinases (MMPs), cathepsins and inflammatory cytokines [22, 23], and indirectly on bone via regulation of monocyte to osteoclast differentiation [24]. In the early RA joint, activated synovial fibroblasts attach to and overgrow the cartilage surface, then invade and destroy cartilage and induce bone resorption (fig. 3). Active degradation of cartilage in vivohas been elegantly demonstrated in the SCID mouse model of arthritis, in whichin vitrocultured RASFs (but not normal or osteoarthritis synovial fibroblasts) attach to and invade co-implanted healthy human cartilage in the absence of cells of the immune system, indicating that this invasive phenotype is both stable and disease specific [25]. Furthermore, RASFs maintain this destructive phenotype even after multiple in vitro passages, and their in vitroinvasiveness correlates with clinical rates of bone erosion in individual patients [26].
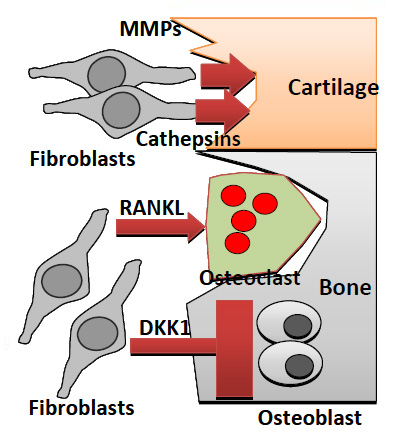
Figure 3
Fibroblast contribute to joint damage. Pannus tissue consists of activated RASFs that attach to and damage cartilage through production of MMPs and cathepsins. In addition, fibroblasts secrete RANKL, which promotes osteoclast differentiation and activation, leading to bone erosions. Furthermore production of DKK-1 inhibits Wnt signaling pathways that normally promote anabolic osteoblast activity, preventing repair of bone erosions. Abbreviations: MMPs: matrix metalloprotenaises; RANKL: receptor activator of nuclear factor k B ligand; DKK-1: dickkopf-1.
It is therefore clear that activated RASFs are not passive players in immune responses but key effectors in the pathogenesis of RA. One of the questions arising from these observations is how these cells become activated and how this activated phenotype persists.
It has been proposed that activation arises from molecular cross-talk between RASFs and other cells of the synovial microenvironment early in disease. Pro-inflammatory cytokines of the initial rheumatoid microenvironment such as TNFα and IL-1 play a key role in the activation of RASFs [23]. Once activated, RASFs produce TNFα, IL-1 and IL-6 that are involved in sustaining regulatory feedback loops and induce production of MMPs, cathepsins, aggrecanases and their inhibitors [6]. However, independently of the fact that pro-inflammatory cytokines induce fibroblast activation, the fact that not all fibroblasts in a joint become activated suggests that there must be other mechanisms in play (intrinsic to these cells) that make them “permissive” to such activation. Indeed, it has conclusively been shown that the persistence of the activated RASFs phenotype is independent of the pro-inflammatory cytokines that initiated the process [25] This may also explain the clinical observation that cessation of anti cytokine therapy (e.g., anti TNF therapy) in the context of clinical RA leads to disease flare [10].
So, what could these intrinsic mechanisms be? Cellular DNA and the epigenetic machinery that regulates its expression are the most plausible candidates to explain unique cellular characteristics.
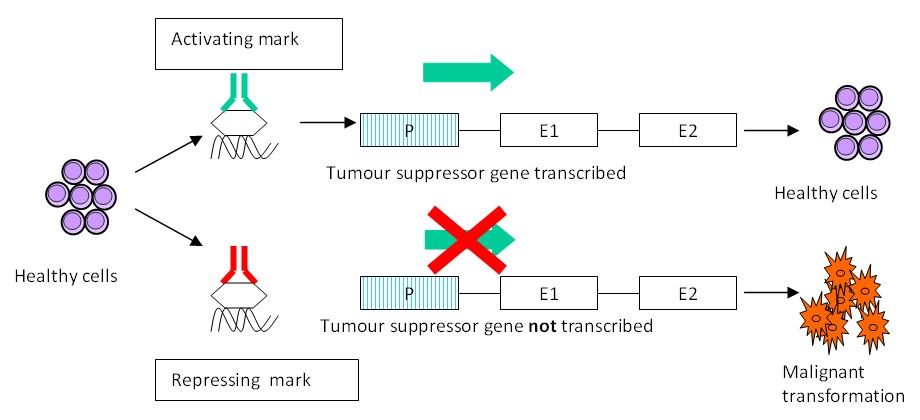
Figure 4
Schematic representation of epigenetics. Epigenetic changes mark the genome at specific sites acting as switches that determine whether a particular gene is transcribed (switch “on”) or not (switch “off”). In the figure an activating mark results in normal transcription of a tumour suppressor gene and maintenance of normal cell phenotype whilst a repressing mark in the same gene results in lack of transcription and malignant transformation of that cell line. Abbreviations: P: promoter; E: exon.
Several lines of evidence indicate that responses to pro-inflammatory stimuli result from persistent changes in the transcription levels of disease relevant genes. RASFs show upregulated expression of proto-oncogenes (c-myc) [27], adhesion molecules (VLA-3, VLA-4, VLA-5) [28] and MMPs (MMP13, MT-MMP) [29, 30] whilst displaying low expression of tumor suppressor genes such as PTEN and p53 [31]. These persistent changes in gene transcription are ultimately responsible for the aggressive behaviour of RASFs. Differential gene expression patterns are not only apparent in disease but also in health and indeed fibroblasts from different anatomical regions have been shown to display characteristic transcriptional profiles that are maintained in vitro[32]. The HOX genes encode the important transcription factors that confer such region-specific identity to tissues [32–34]. But how is this differential gene expression pattern obtained and regulated to provide persistent changes in phenotype? The field of epigenetics explains such regulation, and is increasingly implicated as the determinant of the RASFs phenotype.
Epigenetic changes are hereditable changes in gene expression that do not result from alteration of the underlying DNA sequence [35]. Figure 4 provides a schematic representation of epigenetic mechanisms. The most studied epigenetic modifications to date include non-coding RNA species, DNA methylation and histone modifications. These modifications can be thought of as chemical “switches” that regulate gene expression determining whether a particular gene is expressed (“on”) or not (“off”). In healthy fibroblasts, expression of a non-coding RNA (termed HOTAIR) residing in the HOXC locus results in silencing of genes in the HOXD locus, explaining epigenetic control of HOX gene expression in health [36]. In RA, RASFs display global genomic DNA hypomethylation (an epigenetic change associated with up-regulation of gene expression) that causes up-regulation of disease relevant genes (growth factors, adhesion molecules and MMPs) [37]. Most notably, treatment of normal synovial fibroblasts with the DNA methylation inhibitor 5-aza-2′deoxycytidine (decitabidine) resulted in their transformation into cells resembling activated RASFs [37]. Although this evidence lends support to epigenetic changes as determinants of the RASFs persistent phenotype, further research in this area is needed to unequivocally demonstrate a significant role in the pathogenesis of disease.
Perhaps more noteworthy is that the epigenetic hypothesis may not only provide a mechanistic explanation for this phenotype but a tool to modify it. Most epigenetic modifications are reversible processes catalysed by specific enzymes and cofactors that allow cells to change their gene expression patterns in response to various stimuli. The enzymes and molecular complexes involved in these processes represent very attractive targets for future therapies. As a result, much interest is currently being devoted to this area.
The importance of the stromal microenvironment in disease has been recognised in the cancer field for years. It is now widely accepted that cancer develops as a result of genetic and epigenetic alterations in clonal cells, but that the growth, survival and metastasis of these cells are regulated by stromal-cancer cell interactions [38]. Studies in human lung, breast, colon and prostate cancer have unequivocally demonstrated a key role for stromal cells (so-called cancer associated fibroblasts (CAFs)) in initiation and progression of disease [38]. The term “cancer stroma” has been coined to refer to macroscopic and functional differences between this and the corresponding normal tissue stroma. Cancer stroma is characterized by a modified ECM composition, increased microvessel density, inflammatory cells and activated fibroblasts [39]. Autocrine and paracrine mechanisms resulting in secretion of cytokines, chemokines and growth factors by the stromal microenvironment have been implicated in activation and transformation of fibroblasts into CAFs [40]. Platelet derived growth factor (PDGF) [41] and transforming growth factor beta (TFGβ) are the most important factors in this process. They regulate disease relevant gene expression (fibronectin, PAI-1 and cyclin dependent kinase inhibitors) [42, 43] through stimulation of specific signaling cascades like serine/threonine kinase receptors and the STAT (Signal Transduction and Activation of Transcription) family of signaling pathway regulators. Confirmation of the importance of CAFs comes from invivo studies: co-implantation of pre-malignant prostate epithelial cells with prostate tumour CAFs, but not normal prostate fibroblasts, leads to malignant transformation and proliferation of epithelial cells [44]. In breast cancer, overexpression of TGFβ in mouse fibroblasts induces initiation of breast cancer within normal human epithelium [45].
This compelling evidence demonstrates that tumour-host cross talk at the level of the resident fibroblast cell determines the behaviour of malignant cells. As a result, therapies targeting the tumour stroma are being developed and a number of strategies have been proposed:
1. Targeting signals responsible for activation of fibroblasts: the key role of PDGF and TGFβ signaling pathways has resulted in the development of inhibitors against these molecules. Imatinib (Glivec) inhibits the PDGF-receptor tyrosine kinase and is used for the treatment of chronic myeloid leukaemia and other types of cancer [46]. TGFβ inhibitors are currently in development (e.g., lerdelimumab, metelimumab) [47].
2. Targeting fibroblast specific molecules that initiate and promote tumour growth: an example of this approach is the use a humanised monoclonal antibody (sibrotuzumab) against the fibroblast activation protein (FAP) [48]. FAP is a membrane bound glycoprotein with serine protease activity. It is highly expressed in the tumour stroma and has been shown to enhance tumour growth in vivo[49]. Efficacy has been shown in phase I studies in colorectal cancer. [50]
3. Eliminating activated CAF subpopulations: efforts are being made to determine the differential transcriptional profile of normal fibroblasts compared to CAFs in a variety of malignancies in order to identify unique transcriptional signatures. This will then be further investigated in order to develop new therapies aimed towards elimination of CAFs.
4. Epigenetic therapies. Although still in its early days, research has shown that epigenetic modifications determine the phenotype of cancer and tumour stroma cells promoting their invasiveness and survival. CAFs in human gastric carcinomas exhibit global DNA hypomethylation [51]. More broadly, cancerous epithelial cells are characterized on the one hand by global DNA hypomethylation that promotes chromosomal instability and activates proto-oncogenes, and on the other by regional promoter hypermethylation of specific genes that results in down-regulation of tumour suppressor genes [52]. The understanding of the molecular basis of these epigenetic modifications has led to the development of epigenetic therapies. The DNA methylation inhibitor 5-aza-2′deoxycytidine is currently used for the treatment of myelodysplastic syndromes (MDS), acting by reversing pathological hypermethylation (and hence increasing expression) of the tumour suppressor gene CDKN2B in affected cells [52].
As the understanding of global epigenetic mechanisms in cancer advances, a combinatorial approach using different epigenetic approaches along with standard chemotherapy holds significant promise for effective future treatment. For example, it has been shown that treatment of MDS with 5-aza-2′deoxycytidine is limited by a short duration of response that is improved upon use of combination chemotherapy [53].
Obvious parallels can be drawn between CAFs and RASFs, and indeed current RA research is focusing increasingly upon targeting stromal cells therapeutically.
1. Cytokine blockade. Existing strategies in RA have indirectly targeted stromal cells either by targeting cytokines which impact upon fibroblasts such as anti-TNF therapies (infliximab, etanercept, adalimumab) or by blocking their significant products (anti-IL-6 receptor, tocilizumab) therapies respectively. These therapies share the shortcomings of existing biologics, inhibiting key pleiotropic proinflammatory cytokines with widespread generic roles, leading on the one hand to significant adverse events despite submaximal efficacy, while on the other hand failing to address persistence of disease.
2. Cell signalling pathways. Cellular MAPKinase and NF-κB signalling pathways in particular are heavily implicated in the pro-inflammatory and cartilage damaging activities of synovial fibroblasts. However, the consequences of inhibiting such generic pathways in terms of off-target effects has been likened to opening Pandora’s box [54]. As described in the field of cancer medicine, the approach of targeting more specific upstream activators of pathologically altered fibroblasts via inhibitors of tyrosine kinase family members may lead to fewer off-target effects; imatinib inhibits a narrow family of tyrosine kinases including the PDGF receptor, c-fms and c-kit. It shows promise as a therapy in RA that has been shown to modulate synovial fibroblast proliferation rates [55, 56].
1. Novel fibroblast markers. Historically, the study of fibroblasts has been hampered by a lack of specific cell surface markers. As a result, they have traditionally been identified by their spindle-shaped morphology, elaboration of ECM and lack of markers associated with endothelial, epithelial or haematopoietic cells. This situation is now rapidly changing; an early discovery in this field revealed that the lining layer of synovial fibroblasts is associated with specific expression of cadherin-11, an adhesion molecule responsible for homotypic adhesion between lining layer stromal cells which self organise in the absence of a basement membrane [57]. Not only does this marker co-localise with cells of the lining layer, but in cadherin-11 knockout mice subjected to models of inflammatory arthritis, damage to the joint cartilage is ameliorated [58]. Although not a realistic therapeutic target in isolation, this provided proof of concept that deletion of a stromal sub-population marker could impact on progression of animal models of disease. Further synovial stromal markers have been identified in the rheumatoid synovium with strong overlap with CAF markers. These include FAP, and the sub-lining marker CD248 (endosialin), with restricted stromal expression, and important roles in cancer progression, linkage of hypoxia to angiogenesis, and stromal responses to inflammation [59–61]. Maia et al. have shown that full knockouts and cytoplasmic tail mutants of CD248 result in amelioration of murine arthritis induced by anti-collagen antibodies, offering potential as a therapeutic target [62]. Other specific markers include PBEF (Visfatin), an adipokine with expression closely linked to RA disease activity [63], and FAP, which is expressed in the lining layer of the pathological rheumatoid joint. Inhibitors exist which specifically inhibit both molecules, though exploring their use in arthritis is at an early stage [64, 65].
2. Epigenetic therapies. In vitroand in vivo studies of the use of epigenetic therapies in arthritis have started to emerge. Histone acetylation/decacetylation is an epigenetic modification that has a critical role in regulation of gene transcription by altering chromatin structure. Jüngel and colleagues [66] treated RASFs with the histone deacetylase inhibitor Trichostatin A (TSA). Treatment resulted in inhibition of their proliferation and sensitisation to tumour necrosis factor related apoptosis-inducing ligand (TRAIL) induced apoptosis. Subsequently, Nasu and colleagues [67] conducted an elegant in vivo study to analyse the effects of TSA treatment in the collagen antibody induced arthritis (CAIA) mouse model. Mice were given subcutaneous injections of TSA and histological analysis showed amelioration of clinical arthritis in a dose dependent manner. These studies provide proof of concept that epigenetic therapies may become a reality for the treatment of arthritis. Non-coding RNA species such as microRNAs (miRs) are also becoming rapidly recognised as relatively specific controllers of aberrant gene expression [68]. MiR expression is itself frequently under epigenetic control, highlighting a central mechanism in cancer pathology. One such example in arthritis is miR203. This molecule is under the direct control of DNA methylation, and drives expression of IL-6 and MMP production by synovial fibroblasts. Not only are levels of miR203 raised in longstanding RA compared to osteoarthritis and normal synovial fibroblasts, but expression in the earliest phases of RA when disease is not fully differentiated occurs at an intermediate level, suggesting that the epigenetic control of synovial fibroblast behaviour remains plastic in the earliest phases of disease [69].
At present, experimental approaches to epigenetic modifications have used broad specificity drugs such as TSA and 5-aza-2′deoxycytidine. However, as discussed above, pathological epigenetic changes in carcinoma cells involve complex combinations of gene activation and repression. These therapies do not therefore represent a final goal for rational drug design. The potential for targeted therapy is made possible by the complexes that effectively “read” and “write” the epigenetic code via DNA methylation and a multitude of histone modifications. These complexes, such as the acetyl-lysine reading bromodomains [70] demonstrate high specificity for certain modifications and groups of genes. Engineering of specific antagonists to active sites in these complexes can radically alter disease, as in the case of a malignancy inducing translocation treated by Filippakopoulos et al. who generated a specific cell permeable competitive inhibitor that prevented binding of the BET bromodomain member BRD4, forcing differentiation of a previously incurable cancer [71].
Synovial fibroblasts are key effectors in the pathogenesis of RA. Aspects of the mechanisms involved in the triggering of their abnormal phenotype have begun to be deciphered. Drugs aimed at these cells either via targeting of markers for specific pathologically important subpopulations or modulating epigenetic programming of fibroblasts may be added to the therapeutic arsenal in RA in the near future. Just as in the field of cancer, combinations of epigenetic and conventional antirheumatic therapies may both improve efficacy and modify disease persistence.
1 Scott DL, Pugner K, Kaarela K, Doyle DV, Woolf A, Holmes J, et al. The links between joint damage and disability in rheumatoid arthritis. Rheumatology. (Oxford) 2000;39:122–32.
2 Kitas GD, Erb N. Tackling ischaemic heart disease in rheumatoid arthritis. Rheumatology. (Oxford) 2003;42:607–13.
3 Buckley CD, Pilling D, Lord JM, Akbar AN, Scheel-Toellner D, Salmon M. Fibroblasts regulate the switch from acute resolving to chronic persistent inflammation. Trends Immunol. 2001;22:199–204.
4 Parsonage G, Filer AD, Haworth O, Nash GB, Rainger GE, Salmon M, et al. A stromal address code defined by fibroblasts. Trends Immunol. 2005;26:150–6.
5 Burman A, Haworth O, Hardie DL, Amft EN, Siewert C, Jackson DG, et al. A chemokine-dependent stromal induction mechanism for aberrant lymphocyte accumulation and compromised lymphatic return in rheumatoid arthritis. J Immunol. 2005;174:1693–700.
6 McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol. 2007;7:429–42.
7 McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365:2205–19.
8 van der Helm-van Mil AH, Wesoly JZ, Huizinga TW. Understanding the genetic contribution to rheumatoid arthritis. Curr Opin Rheumatol. 2005;17:299–304.
9 Klareskog L, Padyukov L, Alfredsson L. Smoking as a trigger for inflammatory rheumatic diseases. Curr Opin Rheumatol. 2007;19:49–54.
10 Firestein GS. Evolving concepts of rheumatoid arthritis. Nature. 2003;423:356–61.
11 Takemura S, Braun A, Crowson C, Kurtin PJ, Cofield RH, O’Fallon WM, et al. Lymphoid neogenesis in rheumatoid synovitis. J Immunol. 2001;167:1072–80.
12 Taylor PC, Sivakumar B. Hypoxia and angiogenesis in rheumatoid arthritis. Curr Opin Rheumatol. 2005;17:293–8.
13 Szekanecz Z, Kim J, Koch AE. Chemokines and chemokine receptors in rheumatoid arthritis. Semin Immunol. 2003;15:15–21.
14 Iwamoto T, Okamoto H, Toyama Y, Momohara S. Molecular aspects of rheumatoid arthritis: chemokines in the joints of patients. FEBS J. 2008;275:4448–55.
15 Buckley CD, Amft N, Bradfield PF, Pilling D, Ross E, Arenzana-Seisdedos F, et al. Persistent induction of the chemokine receptor CXCR4 by TGF-beta 1 on synovial T cells contributes to their accumulation within the rheumatoid synovium. J Immunol. 2000;165:3423–9.
16 Bradfield PF, Amft N, Vernon-Wilson E, Exley AE, Parsonage G, Rainger GE, et al. Rheumatoid fibroblast-like synoviocytes overexpress the chemokine stromal cell-derived factor 1 (CXCL12), which supports distinct patterns and rates of CD4+ and CD8+ T cell migration within synovial tissue. Arthritis Rheum. 2003;48:2472–82.
17 Buckley CD. Michael Mason prize essay 2003. Why do leucocytes accumulate within chronically inflamed joints? Rheumatology. (Oxford) 2003;42:1433–44.
18 Ohata J, Zvaifler NJ, Nishio M, Boyle DL, Kalled SL, Carson DA, et al. Fibroblast-like synoviocytes of mesenchymal origin express functional B cell-activating factor of the TNF family in response to proinflammatory cytokines. J Immunol. 2005;174:864–70.
19 Pilling D, Akbar AN, Girdlestone J, Orteu CH, Borthwick NJ, Amft N, et al. Interferon-beta mediates stromal cell rescue of T cells from apoptosis. Eur J Immunol. 1999;29:1041–50.
20 Filer A, Parsonage G, Smith E, Osborne C, Thomas AM, Curnow SJ, et al. Differential survival of leukocyte subsets mediated by synovial, bone marrow, and skin fibroblasts: site-specific versus activation-dependent survival of T cells and neutrophils. Arthritis Rheum. 2006;54:2096–108.
21 Burger JA, Zvaifler NJ, Tsukada N, Firestein GS, Kipps TJ. Fibroblast-like synoviocytes support B-cell pseudoemperipolesis via a stromal cell-derived factor-1- and CD106 (VCAM-1)-dependent mechanism. J Clin Invest. 2001;107:305–15.
22 Muller-Ladner U, Gay S. MMPs and rheumatoid synovial fibroblasts: Siamese twins in joint destruction? Ann Rheum Dis. 2002;61:957–9.
23 Pap T, Muller-Ladner U, Gay RE, Gay S. Fibroblast biology. Role of synovial fibroblasts in the pathogenesis of rheumatoid arthritis. Arthritis Res. 2000;2:361–7.
24 Takayanagi H, Iizuka H, Juji T, Nakagawa T, Yamamoto A, Miyazaki T, et al. Involvement of receptor activator of nuclear factor kappaB ligand/osteoclast differentiation factor in osteoclastogenesis from synoviocytes in rheumatoid arthritis. Arthritis Rheum. 2000;43:259–69.
25 Muller-Ladner U, Kriegsmann J, Franklin BN, Matsumoto S, Geiler T, Gay RE, et al. Synovial fibroblasts of patients with rheumatoid arthritis attach to and invade normal human cartilage when engrafted into SCID mice. Am J Pathol. 1996;149:1607–15.
26 Tolboom TC, van der Helm-van Mil AH, Nelissen RG, Breedveld FC, Toes RE, Huizinga TW. Invasiveness of fibroblast-like synoviocytes is an individual patient characteristic associated with the rate of joint destruction in patients with rheumatoid arthritis. Arthritis Rheum. 2005;52:1999–2002.
27 Qu Z, Garcia CH, O'Rourke LM, Planck SR, Kohli M, Rosenbaum JT. Local proliferation of fibroblast-like synoviocytes contributes to synovial hyperplasia. Results of proliferating cell nuclear antigen/cyclin, c-myc, and nucleolar organizer region staining. Arthritis Rheum. 1994;37:212–20.
28 Rinaldi N, Schwarz-Eywill M, Weis D, Leppelmann-Jansen P, Lukoschek M, Keilholz U, et al. Increased expression of integrins on fibroblast-like synoviocytes from rheumatoid arthritis in vitro correlates with enhanced binding to extracellular matrix proteins. Ann Rheum Dis. 1997;56:45–51.
29 Westhoff CS, Freudiger D, Petrow P, Seyfert C, Zacher J, Kriegsmann J, et al. Characterization of collagenase 3 (matrix metalloproteinase 13) messenger RNA expression in the synovial membrane and synovial fibroblasts of patients with rheumatoid arthritis. Arthritis Rheum. 1999;42:1517–27.
30 Pap T, Shigeyama Y, Kuchen S, Fernihough JK, Simmen B, Gay RE, et al. Differential expression pattern of membrane-type matrix metalloproteinases in rheumatoid arthritis. Arthritis Rheum. 2000;43:1226–32.
31 Pap T, Franz JK, Hummel KM, Jeisy E, Gay R, Gay S. Activation of synovial fibroblasts in rheumatoid arthritis: lack of Expression of the tumour suppressor PTEN at sites of invasive growth and destruction. Arthritis Res. 2000;2:59–64.
32 Chang HY, Chi JT, Dudoit S, Bondre C, van de Rijn M, Botstein D, et al. Diversity, topographic differentiation, and positional memory in human fibroblasts. Proc Natl Acad Sci. USA 2002;99:12877–82.
33 Krumlauf R. Hox genes in vertebrate development. Cell. 1994;78:191–201.
34 Tabin CJ. Why we have (only) five fingers per hand: hox genes and the evolution of paired limbs. Development. 1992;116:289–96.
35 Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21.
36 Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007;129:1311–23.
37 Karouzakis E, Gay RE, Michel BA, Gay S, Neidhart M. DNA hypomethylation in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum 2009;60:3613-22.
38 Micke P, Ostman A. Tumour-stroma interaction: cancer-associated fibroblasts as novel targets in anti-cancer therapy? Lung Cancer. 2004;45(Suppl 2):S163–S175.
39 De WO, Mareel M. Role of tissue stroma in cancer cell invasion. J Pathol. 2003;200:429–47.
40 Powell DW, Mifflin RC, Valentich JD, Crowe SE, Saada JI, West AB. Myofibroblasts. I. Paracrine cells important in health and disease. Am J Physiol. 1999;277:C1–C9.
41 Elenbaas B, Weinberg RA. Heterotypic signaling between epithelial tumor cells and fibroblasts in carcinoma formation. Exp Cell Res. 2001;264:169–84.
42 Moustakas A, Souchelnytskyi S, Heldin CH. Smad regulation in TGF-beta signal transduction. J Cell Sci. 2001;114:4359–69.
43 Piek E, Heldin CH, Ten DP. Specificity, diversity, and regulation in TGF-beta superfamily signaling. FASEB J. 1999;13:2105–24.
44 Olumi AF, Grossfeld GD, Hayward SW, Carroll PR, Tlsty TD, Cunha GR. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999;59:5002–11.
45 Kuperwasser C, Chavarria T, Wu M, Magrane G, Gray JW, Carey L, et al. Reconstruction of functionally normal and malignant human breast tissues in mice. Proc Natl Acad Sci U S A. 2004;101:4966–71.
46 Capdeville R, Buchdunger E, Zimmermann J, Matter A. Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat Rev Drug Discov. 2002;1:493–502.
47 Bonafoux D, Lee WC. Strategies for TGF-beta modulation: a review of recent patents. Expert Opin Ther Pat. 2009;19:1759–69.
48 Scott AM, Wiseman G, Welt S, Adjei A, Lee FT, Hopkins W, et al. A Phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clin Cancer Res. 2003;9:1639–47.
49 Cheng JD, Dunbrack RL, Jr., Valianou M, Rogatko A, Alpaugh RK, Weiner LM. Promotion of tumor growth by murine fibroblast activation protein, a serine protease, in an animal model. Cancer Res. 2002;62:4767–72.
50 Welt S, Divgi CR, Scott AM, Garin-Chesa P, Finn RD, Graham M, et al. Antibody targeting in metastatic colon cancer: a phase I study of monoclonal antibody F19 against a cell-surface protein of reactive tumor stromal fibroblasts. J Clin Oncol. 1994;12:1193–203.
51 Jiang L, Gonda TA, Gamble MV, Salas M, Seshan V, Tu S, et al. Global hypomethylation of genomic DNA in cancer-associated myofibroblasts. Cancer Res. 2008;68:9900–8.
52 Raj K, Mufti GJ. Azacytidine (Vidaza(R)) in the treatment of myelodysplastic syndromes. Ther Clin Risk Manag. 2006;2:377–88.
53 Silverman LR, Demakos EP, Peterson BL, Kornblith AB, Holland JC, Odchimar-Reissig R, et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol. 2002;20:2429–40.
54 Wendling D, Prati C, Toussirot E, Herbein G. Targeting intracellular signaling pathways to treat rheumatoid arthritis: Pandora's box? Joint Bone Spine. 2010;77:96–8.
55 Paniagua RT, Sharpe O, Ho PP, Chan SM, Chang A, Higgins JP, et al. Selective tyrosine kinase inhibition by imatinib mesylate for the treatment of autoimmune arthritis. J Clin Invest. 2006;116:2633–42.
56 Terabe F, Kitano M, Kawai M, Kuwahara Y, Hirano T, Arimitsu J, et al. Imatinib mesylate inhibited rat adjuvant arthritis and PDGF-dependent growth of synovial fibroblast via interference with the Akt signaling pathway. Mod Rheumatol. 2009;19:522–9.
57 Valencia X, Higgins JM, Kiener HP, Lee DM, Podrebarac TA, Dascher CC, et al. Cadherin-11 provides specific cellular adhesion between fibroblast-like synoviocytes. J Exp Med. 2004;200:1673–9.
58 Lee DM, Kiener HP, Agarwal SK, Noss EH, Watts GF, Chisaka O, et al. Cadherin-11 in synovial lining formation and pathology in arthritis. Science. 2007;315:1006–10.
59 MacFadyen JR, Haworth O, Roberston D, Hardie D, Webster MT, Morris HR, et al. Endosialin (TEM1, CD248) is a marker of stromal fibroblasts and is not selectively expressed on tumour endothelium. FEBS Lett. 2005;579:2569–75.
60 Lax S, Hou TZ, Jenkinson E, Salmon M, MacFadyen JR, Isacke CM, et al. CD248/Endosialin is dynamically expressed on a subset of stromal cells during lymphoid tissue development, splenic remodeling and repair. FEBS Lett. 2007;581:3550–6.
61 Tomkowicz B, Rybinski K, Sebeck D, Sass P, Nicolaides NC, Grasso L, et al. Endosialin/TEM-1/CD248 regulates pericyte proliferation through PDGF receptor signaling. Cancer Biol Ther. 2010;9:908–15.
62 Maia M, de VA, Janssens T, Moons M, van LK, Tavernier J, et al. CD248 and its cytoplasmic domain: a therapeutic target for arthritis. Arthritis Rheum. 2010;62:3595–606.
63 Brentano F, Schorr O, Ospelt C, Stanczyk J, Gay RE, Gay S, et al. Pre-B cell colony-enhancing factor/visfatin, a new marker of inflammation in rheumatoid arthritis with proinflammatory and matrix-degrading activities. Arthritis Rheum. 2007;56:2829–39.
64 Evans L, Williams AS, Hayes AJ, Jones SA, Nowell M. Suppression of leukocyte infiltration and cartilage degradation by selective inhibition of pre-B cell colony-enhancing factor/visfatin/nicotinamide phosphoribosyltransferase: Apo866-mediated therapy in human fibroblasts and murine collagen-induced arthritis. Arthritis Rheum. 2011;63:1866–77.
65 Ospelt C, Mertens JC, Jungel A, Brentano F, Maciejewska-Rodriguez H, Huber LC, et al. Inhibition of fibroblast activation protein and dipeptidylpeptidase 4 increases cartilage invasion by rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2010;62:1224–35.
66 Jungel A, Baresova V, Ospelt C, Simmen BR, Michel BA, Gay RE, et al. Trichostatin A sensitises rheumatoid arthritis synovial fibroblasts for TRAIL-induced apoptosis. Ann Rheum Dis. 2006;65:910–2.
67 Nasu Y, Nishida K, Miyazawa S, Komiyama T, Kadota Y, Abe N, et al. Trichostatin A, a histone deacetylase inhibitor, suppresses synovial inflammation and subsequent cartilage destruction in a collagen antibody-induced arthritis mouse model. Osteoarthritis Cartilage. 2008;16:723–32.
68 Stefani G, Slack FJ. Small non-coding RNAs in animal development. Nat Rev Mol Cell Biol. 2008;9:219–30.
69 Stanczyk J, Ospelt C, Karouzakis E, Filer A, Raza K, Kolling C, et al. Altered expression of microRNA-203 in rheumatoid arthritis synovial fibroblasts and its role in fibroblast activation. Arthritis Rheum. 2011;63:373–81.
70 Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28:1057–68.
71 Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–73.
Funding / potential competing interests: Funding for this work has been provided by Wellcome Trust, ARUK, and AutoCure.