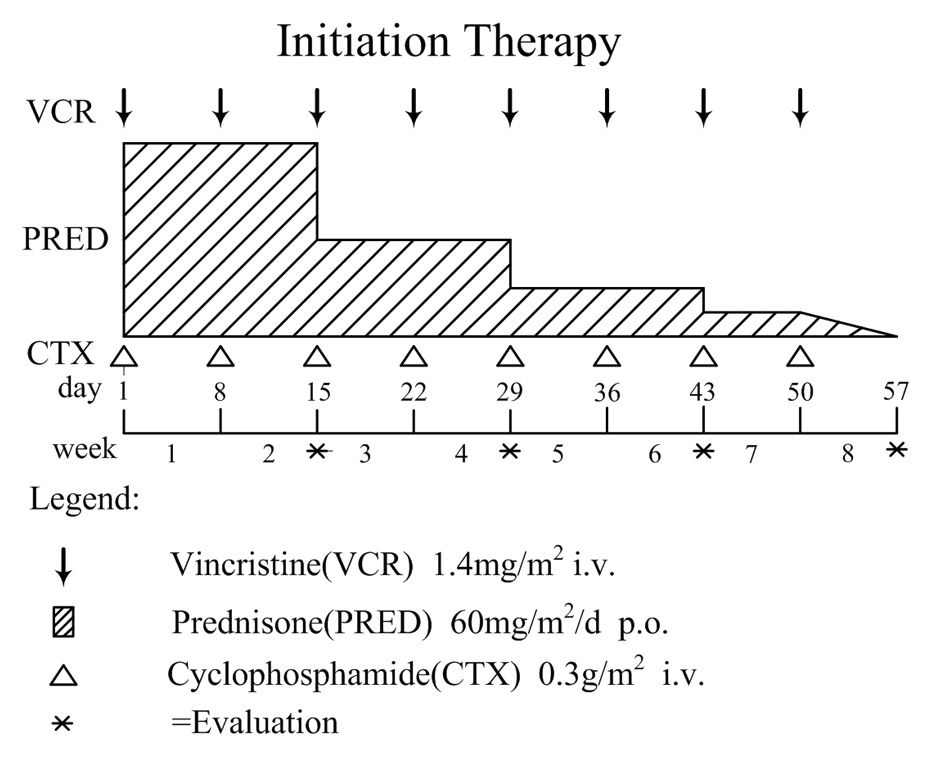
Figure 1A
Overview of initial treatment protocol for secondary hemophagocytic lymphohistiocytosis (sHLH).
DOI: https://doi.org/10.4414/smw.2012.13512
Haemophagocytic lymphohistiocytosis (HLH) is a potentially fatal hyperinflammatory syndrome causing persistent high fever, hepatosplenomegaly, pancytopenia and haemophagocytosis in the bone marrow. Features of cytokine flooding, such as IL-6, IL-12, INF-γ and TNF-α, and also high plasma concentrations of sCD25 and sCD163, correlate with the clinical and laboratory manifestations [1–5]. The annual incidence of HLH is estimated at approximately one case per 800,000 individuals [6]. Broadly, HLH can be classified into either familial or secondary HLH. Familial HLH (FHL) is an autosomal recessive disorder with a median survival of less than two months after diagnosis if untreated, with onset typically during infancy or early childhood [1]. Secondary HLH (sHLH) may occur in association with severe infections, malignancies or autoimmune diseases. Although it may subside spontaneously, it may also be associated with pronounced mortality.
Much effort has been expended on controlling the hyperinflammatory syndrome. Aggressive treatments, such as the HLH-2004 protocol and allogeneic haematopoietic stem-cell transplantation (allo-HSCT) [7, 8], are most strongly recommended. However, they have been designed mainly for primary, inherited FHL, normally in patients aged <18 years, as well as any severe form of HLH. Reports on the treatment of sHLH are diverse, and no standard treatment protocol has been established. Currently, the conservative/mild treatments without etoposide, such as corticosteroid/cyclosporine (CsA), intravenous immunoglobulins (IVIG) and the CHOP regimen, have shown varying effectiveness. The cyclophosphamide, vincristine, and prednisone (COP) regimen is used in many haematological diseases [9, 10].We investigated treatment outcome after its use as another mild immunomodulatory therapy in sHLH patients.
From June 2007 to May 2011, 15 patients diagnosed with HLH were enrolled and treated with the COP regimen as either initial or second-line therapy. HLH was diagnosed according to the revised diagnostic guidelines of the Histiocyte Society’s HLH Study Group [1]. To rule out a possible underlying disease, e.g. virus or bacterial infection, autoimmune disease and neoplasms (especially lymphoma), we selectively and systematically performed a series of laboratory tests, including culturing of blood, bone marrow or sputum; screening of tumour markers and autoantibodies; computed tomography (CT) scans; positron emission tomography (PET)/CT; and in some cases lymph-node biopsy. Finally, 11 of 15 patients were identified as having sHLH. Among them, 7 patients were identified as having infection-associated HLH, 2 patients as having autoimmune disease-associated HLH, and another 2 patients as having malignant lymphoma associated with HLH. No underlying disease was found in the remaining 4 patients.
After diagnosing HLH and investigating any possible underlying disease, we adopted the COP regimen as an initial treatment. The initial therapy included cyclophosphamide (CTX) 0.3 g/m2 i.v. and vincristine (VCR) 1.4 mg/m2 i.v., once weekly for the first eight weeks, and prednisone (PRED) 60 mg/m2/d p.o., for the first 2 weeks, reduced by 50% of the previous dose every 2 weeks until the end of the 7th week, then progressively tapered for 1 week (fig. 1A). CTX and VCR were delayed, provided that the absolute neutrophil count (ANC) was below 0.5×109/L or in the presence of severe myelosuppression. If no improvement of symptoms or laboratory tests was seen on the 14th day of the COP regimen, the patient was switched to an alternative regimen.
Figure 1A
Overview of initial treatment protocol for secondary hemophagocytic lymphohistiocytosis (sHLH).
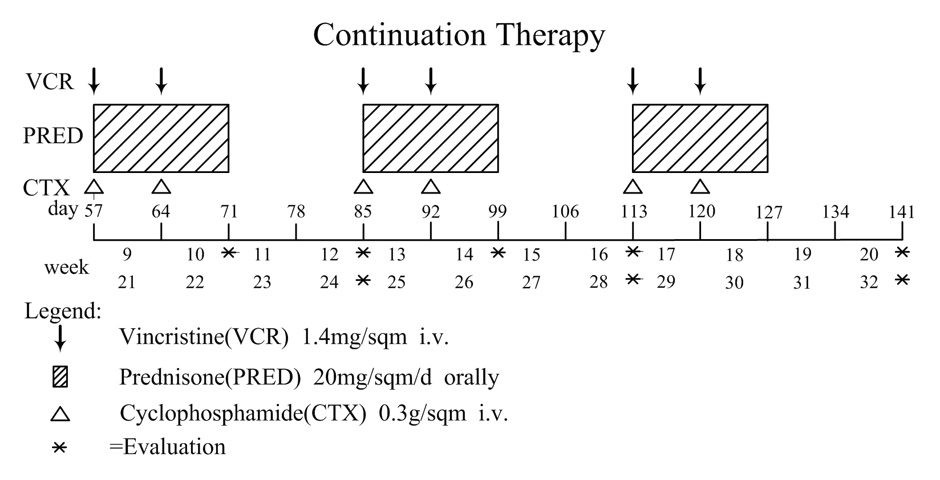
Figure 1B
Overview of continuation treatment protocoI for secondary hemophagocytic lymphohistiocytosis (sHLH).
Continuation therapy was administered to patients who responded but did not achieve a complete response (CR) after the initial therapy. Once CR was achieved during continuation therapy the treatment of HLH patients with the COP regimen was stopped. Patients were treated with COP (CTX 0.3 g/m2 i.v. on day 1, day 8; VCR 1.4 mg/m2 i.v. on day 1, day 8; PRED 20 mg/m2 p.o., for 14 days). Continuation therapy was repeated every 4 weeks for a total of 4 to 6 cycles in responding patients (fig. 1B). The COP regimen was delayed if ANC was less than 0.5×109/L or in the case of severe myelosuppression. If relapse or refractory disease was identified during continuation therapy, the patient was switched to alternative regimens such as the HLH-2004 treatment protocol, IVIG and other clinical trials.
The aim of subsequent therapy was to sustain remission in patients who had achieved CR. If there were some abnormalities in certain parameters but the patient had not met the definition of relapse [3], the following treatment strategies might be considered: 1. continue the continuation therapy; 2. prolong the intervals between every cycle of treatment described in the continuation therapy protocol; or 3. administer prednisone only in the minimum effective dose.
Appropriate broad-spectrum antibiotics were necessary (as determined by culture results), and maximal initial supportive care was suggested. The supportive therapy included prophylactic cotrimoxazole, antimycotic and/or antiviral therapy in suspected patients, and IVIG (during initial and continuation therapy). Supportive transfusion, including red blood cells, platelets, fresh-frozen plasma or cryoprecipitate, was adopted depending on blood cell count, the results of the blood coagulation test and the patient’s clinical status.
Complete response (CR) was defined as disappearance of clinical symptoms and resolution of all signs and laboratory abnormalities. Partial response (PR) was defined as significant but incomplete improvement of clinical and/or biological manifestations. If either of these was aggravated, the response was considered no response (NR). Response was evaluated every two weeks during treatment and at the point of discharge or death. Follow-up, mainly focusing on clinical observation and laboratory detection, was conducted every two weeks for two months and then every month for four months.
A retrospective analysis of the efficacy of COP chemotherapy was performed. Survival time was calculated from the initial treatment to the time of analysis or death. Statistical significance was determined at a level of p <0.05. The Wilcoxon test was used to calculate the difference in parameters before and after chemotherapy. The software package SPSS for Windows (version 16.0, SPSS, Chicago, IL, USA) was used for the statistical analysis.
The study group consisted of fifteen patients (10 females, 5 males) with ages ranging from 14 to 73 years (median 41 years). Two patients had autoimmune diseases. Two patients were classified as having lymphoma-associated HLH (LAHLH), and seven as having infection-associated HLH (IAHLH). Four patients developed the disease in the absence of apparent underlying disease. Table 1 shows the major clinical and laboratory characteristics of 15 patients with HLH at diagnosis. Most of the patients with HLH presented with high fever (100%), splenomegaly (73.3%), lymphadenectasis (46.7%) and bleeding (46.7%). Hepatomegaly, oedema and rash accounted for 33.3%, while neurological symptoms, jaundice and multi-cavity effusions were each observed in three cases (20.0%).
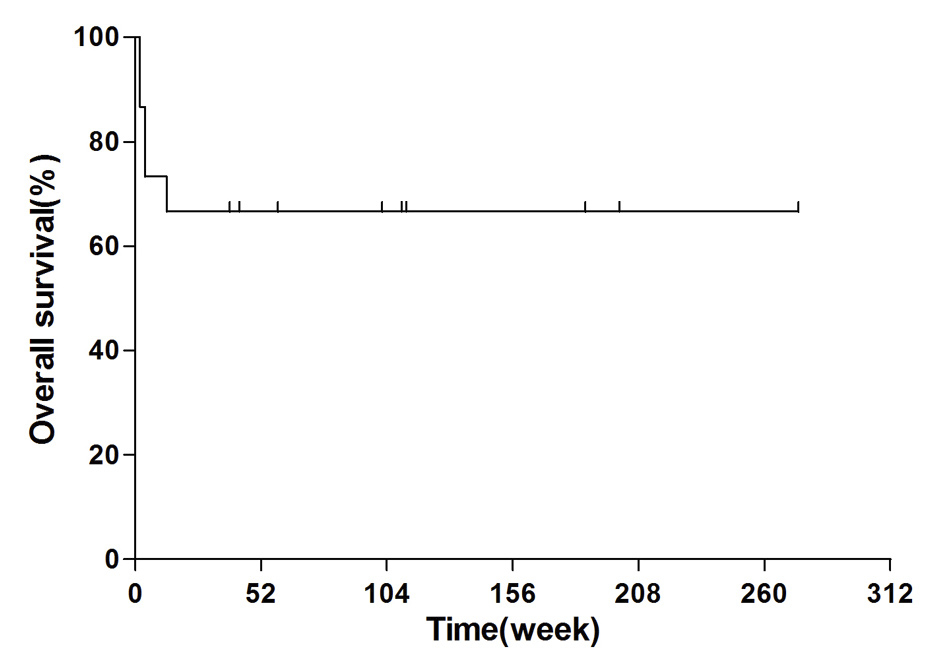
Figure 2
The one-year probability of overall survival of the fifteen patients with sHLH treated with COP regimen was 66.7%.
Bone marrow aspirations were analysed at least once for each patient. Most of them revealed varying degrees of histiocyte proliferation with active haemophagocytosis at the time of diagnosis, except for one patient for whom this condition was detected in her secondary aspiration. Nine of the ten patient samples examined by flow cytometry showed inversion of the CD4+/CD8+ratio and a decrease in the number of natural killer (NK) cells; only one patient showed normal expression.
A median of 4.0 cycles of chemotherapy (CTX plus VCR) were administered to the patients initially, with a range of 1 to 8 times depending on their different aetiological factors and the conditions of the disease. Six out of the fifteen patients (two with autoimmune disease, three with infections, one of doubtful cause) received 6 to 8 cycles. Eight patients (two with non-Hodgkin’s lymphoma [NHL], four with infections and two of unknown aetiology) were given 1 to 4 cycles. Twelve patients did not need continuation therapy, except for three who reached CR at 4 to 6 cycles of continuation therapy. The overall response (OR) rate was 80.0%, consisting of seven CR and five PR. Three patients showed NR to this therapy. All the CR cases are currently alive, having had either long-term treatment or short-term treatment with a mild infection.
We performed a mean follow-up duration of 72.5 weeks (range 2–204 weeks) for each patient until the cut-off time of May 13, 2011. The one-year probability of overall survival (OS) was 10/15 (66.7%) (fig. 2). One patient (case 3) with NHL and another (case 11) with EBV infection showed no response to chemotherapy and died within two weeks. One (case 7, 69 yrs) with MRSA infection obtained PR after 4 episodes of chemotherapy (CTX plus VCR) but had only four weeks’ duration and died after rapid reduction of the prednisone dose. Another (case 9) with CMV infection also succumbed to early reduction of the steroid dose and the resulting severe pneumonia. The last patient who died (case 13) without obvious underlying aetiology showed aggravated disease after one episode of initial chemotherapy. The HLH-2004 treatment protocol was used as a salvage strategy, but the patient still died of severe myelosuppression. The detailed data are shown in tables 2 and 3.
The levels of most laboratory parameters such as neutrophil counts, platelet counts (PLT), serum ferritin (SF), lactate dehydrogenase (LDH) and fibrinogen (FIB) improved significantly after chemotherapy when compared with levels at disease onset (means ± standard deviations, P <0.05, data not shown). However, no significant change in HGB and TG was identified.
All 15 patients were assessed for toxicities associated with the COP regimen. Table IV summarises grade I–III haematological and non-haematological toxicities of the COP regimen administered to 15 patients. Toxicities were predominantly grade I or II, which most patients could tolerate well without requiring dose reduction of the full-dose COP, such that 86.7% of the intended dose was delivered throughout the chemotherapy regimen. One older patient (6.67%) suffered grade-III myelosuppression, and another (6.67%) experienced grade-III pneumonia. The most common non-haematological toxicity was gastrointestinal toxicity, e.g., nausea/vomiting, stomatitis and transient constipation. However, these patients were all well controlled with supportive management and were able to complete the chemotherapy safely. No renal or cardiac toxicity was observed. No patient developed a secondary tumour.
| Table 1: Clinical and laboratory characteristics of 15 patients with HLH at diagnosis. | ||||
| Characteristics | No. (%) of patients | Characteristics | No. (%) of patients | |
| Age, years | 44, range 14–73 | Jaundice | 3 (20.0) | |
| Gender | Laboratory finding | |||
| Male/female | 5 (33.3)/10 (66.7) | PLT (<100×109/L) | 15 (100) | |
| Underlying cause | Neu (<1.0×109/L) | 10 (66.7) | ||
| Infection | 7 (46.7) | Hb (<90 g/L) | 8 (53.3) | |
| Autoimmune disease | 2 (13.3) | SF (≥500 g/L) | 15 (100) | |
| Malignant lymphoma | 2 (13.3) | FIB (<1.5 g/L) | 10 (66.7) | |
| Unknown cause | 4 (26.7) | TG (>3 mmol/L) | 9 (60.0) | |
| Clinical manifestation | Increased ALT/AST | 12 (80.0) | ||
| Persistent fever | 15 (100) | Increased LDH | 14 (93.3) | |
| Splenomegaly | 11 (73.3) | Increased TIBL | 5 (33.3) | |
| Lymphadenectasis | 7 (46.7) | Increased VLDL/Decreased HDL | 10 (66.7) | |
| Bleeding | 7 (46.7) | Prolonged PT/APTT | 8 (53.3) | |
| Hepatomegaly | 5 (33.3) | Hyponatremia | 8 (53.3) | |
| Oedema | 5 (33.3) | Hypoproteinemia | 11 (73.3) | |
| Rash | 5 (33.3) | BM hemophagocytosis | 14 (93.3) | |
| Multicavity effusion | 3 (20.0) | |||
| Neurological symptom | 3 (20.0) | |||
| PLT, platelets; Neu, neutrophil; Hb, haemoglobin; SF, serum ferritin; FIB, fibrinogen; TG, triglyceride; ALT, alanine aminotransferase; AST, aspartate aminotransferase; LDH, lactate dehydrogenase; TBIL, total bilirubin; VLDL, very low-density lipoprotein; HDL, high-density lipoprotein; PT, prothrombin time; APTT, activated partial thromboplastin time; BM, bone marrow. | ||||
| Table 2: Detailed treatment course and prognosis. | ||||||||
| NO. | Gender/Age | Aetiology | Chemotherapy (VCR+CTX) times of initial therapy | Continuation therapy cycle | Treatment effect | Survival time (W) | Salvage | Prognosis |
| 1 | Female/14 | Autoimmune disease | 6 | CR | 204 | no | Alive | |
| 2 | Female/38 | Autoimmune disease | 8 | 12 | CR | 59 | no | Alive |
| 3 | Female/41 | Malignant lymphoma | 2 | NR | 2 | no | Death | |
| 4 | Male/42 | Malignant lymphoma | 3 | PR | 186 | allo-HSCT | Alive | |
| 5 | Female/19 | Infection (MRSA) | 2 | CR | 110 | no | Alive | |
| 6 | Female/27 | Infection (CMV) | 8 | 8 | CR | 112 | no | Alive |
| 7 | Female/69 | Infection (MRSA) | 4 | PR | 4 | no | Death | |
| 8 | Male/32 | Infection (bacteria) | 2 | CR | 164 | no | Alive | |
| 9 | Female/25 | Infection (CMV) | 7 | PR | 13 | no | Death | |
| 10 | Female/63 | Infection (bacteria) | 8 | 8 | CR | 39 | no | Alive |
| 11 | Female/56 | Infection (EBV) | 1 | NR | 2 | HLH-2004 | Death | |
| 12 | Male/69 | Unknown cause | 6 | CR | 102 | no | Alive | |
| 13 | Female/53 | Unknown cause | 1 | NR | 4 | HLH-2004 | Death | |
| 14 | Male/40 | Unknown cause | 2 | PR | 43 | no | Alive | |
| 15 | Male/73 | Unknown cause | 5 | PR | 43 | no | Alive | |
| MRSA, methicillin resistant staphylococcus aureus; EBV, Epstein-Barr virus; CMV, cytomegalovirus; allo-HSCT, allogene haematopoietic stem-cell transplantation. | ||||||||
| Table 3: Response rate of HLH depending on aetiology. | ||||
| Aetiology | Response rate | |||
| Total | CR | PR | NR | |
| Total | 15 | 7 (46.7%) | 5 (33.3%) | 3 (20.0%) |
| Autoimmune disease | 2 | 2 (100.0%) | 0 (0.0%) | 0 (0.0%) |
| Malignant lymphoma | 2 | 0 (0.0%) | 1 (50.0%) | 1 (50.0%) |
| Infection | 7 | 4 (57.1%) | 2 (28.6%) | 1 (14.3%) |
| Unknown cause | 4 | 1 (25.0%) | 2 (50.0%) | 1 (25.0%) |
| CR, complete response; PR, partial response; NR, no response. | ||||
| Table 4: Toxicities associated with COP chemotherapy. | |||
| Toxicity | Grade (WHO) | ||
| I | II | III | |
| Anaemia | 5 (33.3%) | 3 (20.0%) | 0 (0.0%) |
| Neutropenia | 3 (20.0%) | 2 (13.3%) | 1 (6.67%) |
| Thrombocytopenia | 3 (20.0%) | 1 (6.67%) | 1 (6.67%) |
| Infection | 4 (26.7%) | 3 (20.0%) | 1 (6.67%) |
| Nausea/vomiting/stomatitis | 5 (33.3%) | 4 (26.7%) | 0 (0.0%) |
| Constipation | 4 (26.7%) | 1 (6.67%) | 0 (0.0%) |
| Peripheral neurotoxicity | 1 (6.67%) | 0 (0.00%) | 0 (0.0%) |
| Alopecia | 3 (20.0%) | 0 (0.00%) | 0 (0.0%) |
Though we enrolled these patients according to the diagnostic criteria of the HLH-2004 protocol, all were consistent with the new diagnostic criteria proposed by the American Society of Hematology (ASH), 2009 [11]. Hepatitis was added to the criteria for the first time, while hypertriglyceridaemia and hypofibrinogenaemia were downgraded as auxiliary rather than primary results. In our study, 80.0% of patients displayed ALT/AST increases, while 66.7% presented hypofibrinogenaemia, and only 60.0% hypertriglyceridaemia, apparently offering more evidence in support of the new criteria. The hepatitis was mild and sensitive to our treatment: a significant reduction of ALT levels was found after chemotherapy (P = 0.019). In terms of reflecting the efficacy of the COP regimen, the SF, neutrophilic granulocyte, PLT and serum LDH levels were confirmed by assessment values. However, we did not observe any significant change in the TG level, which has been suggested as a surrogate marker for diagnosis of HLH and evaluation of treatment response [12].
In our study we witnessed an encouraging outcome after treating patients with the COP regimen. The overall response rate was 80% (CR 46.7% + PR 33.3%), and the one-year probability of OS was 66.7%. A large series of children treated with HLH-94 showed a 75% complete remission rate and 3-year OS of 55% ± 9% [13]. Another survey observed 73% CR and 24% PR with a combination of antithymocyte globulins (ATG) with corticosteroids, cyclosporine A and intrathecal injections of methotrexate [14]. When referring to adults, Shin reported an OR of 58.8% (CR 41.2% + PR 17.6%) and 43.9% two-year OS with the CHOP regimen [15]. Despite differences in the combination of patient series and age distributions, our results are in accordance with these previous studies. Interestingly, patients with different aetiologies showed different responses to the COP regimen. IAHLH is most sensitive to it, especially bacteria-induced cases. A short course is often enough in younger patients. When a relatively longer treatment course was needed in older patients and in virus-associated HLH (VAHLH) other than EBV, IVIG supplementation was helpful. EBV-HLH is associated with high mortality. Patients with EBV infection in our study progressed rapidly and died quickly, a course which could not be controlled through use of the COP regimen. Early institution of etoposide-containing treatment may improve survival [16]. As many as half of the patients with HLH secondary to autoimmune disease may respond to corticosteroids alone [17, 18]. Aggressive supportive management and high-dose corticosteroids were recommended as an initial step by some authors, while CsA, etoposide, IVIG, plasma exchange and allo-HSCT have been suggested as second-line therapies for refractory disease [17, 19]. In our series use of the COP regimen yielded a satisfactory prognosis for the patients with autoimmune disease-associated HLH. Conceivably it suppressed the disturbed immune system, which is the common pathological mechanism of both autoimmune disease and HLH. After use of the COP regimen to reduce disease severity, HLH gradually subsided in some patients with unknown aetiologies. Malignancy-associated HLH (MAHLH) often impedes adequate treatment of malignancy and has the worst outcome compared with any other form of HLH. In our study we adopted the COP regimen to control the existing HLH before malignant lymphoma was certified. One patient showed no response and died rapidly. Another, although PR was achieved, might still have had a poor prognosis if no allo-HSCT had been administered. In our opinion the COP regimen may not be sufficient when treating patients with MAHLH. First-line chemotherapy is of the utmost importance, and allo-HSCT should be considered in eligible individuals, as reported by Balwierz [20]. The COP protocol did relieve the symptoms and laboratory abnormalities of HLH triggered by an activated immune system. However, treatment aimed directly at the aetiology may cure the disease more efficiently. It is therefore important to find the underlying disease causing sHLH. If the COP regimen is administered too early it may conceal aetiological factors and be unfavourable for prognosis, even if the HLH is definite. Combining the COP regimen with aetiological treatment and aggressive supportive therapy seems to work best.
Currently, national multicentre research on the efficacy of the HLH-2004 treatment protocol is underway; however, related reports are rare at present. Zhu reported that 7/14 paediatric patients (50%) who were treated with the HLH-2004 protocol ultimately died; 4/14 relapsed or showed unremitting activation [21]. Patrick observed that 5/17 paediatric patients (29.4%) experienced severe neurological toxicity while being treated with the HLH-2004 treatment protocol [22]. Cardiotoxicity has been reported as a side effect of etoposide, primarily in adults [23]. Seo even reported a case of therapy-related acute monocytic leukaemia following low-dose etoposide treatment for HLH [24]. In our series we treated a 53-year-old patient with relapsed disease using the HLH-2004 treatment protocol as a salvage strategy. Unfortunately the myelosuppression was so severe that she died of an uncontrollable opportunistic infection. In another report of 10 adult cases treated with the recommended HLH-2004 protocol in our department, mortality was as high as 60% [25]. In contrast, common side effects of chemotherapy, such as anorexia, nausea, vomiting, constipation and peripheral neuropathy, were so mild in our study that relief was obtained through appropriate supportive care. Furthermore, myelosuppression was mainly grade I–II without obvious renal or cardiac toxicities. Patients with HLH usually present severe disease status at the time of onset. The COP regimen may be more suitable than other chemotherapy regimens, such as HLH-2004 and CHOP. In HLH patients who presented with bacterial infection or autoimmune disease, or patients who cannot tolerate intensive chemotherapy due to old age or disease severity, the COP regimen may be the first choice for treatment.
In conclusion, COP chemotherapy had a relatively favourable effect for adult patients with sHLH, and the toxicities were tolerable. However, the heterogeneous prognosis depends, to some extent, on different aetiological factors and the choice of treatment. Given the small number of patients included, further studies should enroll more patients.
Acknowledgements: We express appreciation for the assistance given by the clinicians, nurses and laboratory workers at The First Affiliated Hospital of Nanjing Medical University, Jiangsu Province Hospital.
1 Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: Diagnostic and Therapeutic Guidelines for Hemophagocytic Lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124–31.
2 Szyper-Kravitz M. The hemophagocytic syndrome/macrophage activation syndrome: a final common pathway of a cytokine storm. Isr Med Assoc J. 2009;11:633–4.
3 Gupta S, Weitzman S. Primary and secondary hemophagocytic lymphohistiocytosis: clinical features, pathogenesis and therapy. Expert Rev Clin Immunol. 2010; 6:137–54.
4 Haddad E, Sulis ML, Jabado N, Blanche S, Fischer A, Tardieu M. Frequency and severity of central nervous system lesions in hemophagocytic lymphohistiocytosis. Blood. 1997; 89:794–800.
5 Billiau AD, Roskams T, Van Damme-Lombaerts R, Matthys P, Wouters C. Macrophage activation syndrome: characteristic findings on liver biopsy illustrating the key role of activated, IFN-γ-producing lymphocytes and IL-6- and TNF-α-produing macrophage. Blood. 2005;105:1648–51.
6 Ishii E, Ohga S, Imashuku S, Yasukawa M, Tsuda H, Miura I, et al. Nationwide survey of hemophagocytic lymphohistiocytosis in Japan. Int J Hematol. 2007;86:58–65.
7 Blanche S, Caniglia M, Girault D, Landman J, Griscelli C, Fischer A. Treatment of hemophagocytic lymphohistiocytosis with 22 cases of chemotherapy and bone marrow transplantation: a single-center study. Blood. 1991;78:51–4.
8 Cesaro S, Locatelli F, Lanino E, Porta F, Di Maio L, Messina C, et al. Hematopoietic stem cell transplantation for hemophagocytic lymphohistiocytosis: a retrospective analysis of data from the Italian Association of Pediatric Hematology Oncology (AIEOP). Haematologica. 2008;93:1694–701.
9 Marcus R, Imrie K, Belch A, Cunningham D, Flores E, Catalano J, et al. CVP chemotherapy plus rituximab compared with CVP as first-line treatment for advanced follicular lymphoma. Blood. 2005;105:1417–23.
10 Wang SH, Wang QS, Sun L, Li HH, Zhao Y, Jia BJ, et al. Clinical analysis of 12 patients with angioimmunoblastic T cell lymphoma. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2010;18:1208–10.
11 Filipovich AH. Hemophagocytic lymphohistiocytosis (HLH) and related disorders. Hematology Am Soc Hematol Educ Program. 2009:127–31.
12 Okamoto M, Yamaguchi H, Isobe Y, Yokose N, Mizuki T, Tajika K, et al. Analysis of triglyceride value in the diagnosis and treatment response of secondary hemophagocytic syndrome. Intern Med. 2009;48:775–81.
13 Henter JI, Samuelsson Horne A, Arico M, Egeler RM, Elinder G, Filipovich AH, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002;100:2367–73.
14 Nizar Mahlaoui, Marie Ouachée-Chardin, Geneviève de Saint Basile, Bénédicte. Immunotherapy of familial hemophagocytic lymphohistiocytosis with antithymocyte globulins: A single-center retrospective report of 38 patients. Pediatrics. 2007;120:622–8.
15 Shin HJ, Chung JS, Lee JJ, Sohn SK, Choi YJ, Kim YK, et al. Treatment outcomes with CHOP chemotherapy in adult patients with hemophagocytic lymphohistiocytosis. J Korean Med Sci. 2008;23:439–44.
16 Imshuku S, Kuriyama K, Teramura T, Ishii E, Kinugawa N, Kato M, et al. Requirement for etoposide in the treatment of Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis. J Clin Oncol. 2001;19:2665–73.
17 Kelly A, Ramanan AV. Recognition and management of macrophage activation syndrome in juvenile arthritis. Curr Opin Rheumatol. 2007;19:477–81.
18 Stéphan JL, Koné-Paut I, Galambrun C, Mouy R, Bader-Meunier B, Prieur AM. Reactive haemophagocytic syndrome in children with inflammatory disorders: a retrospective study of 24 patients. Rheumatology. (Oxford) 2001;40:1285–92.
19 Emmenegger U, Schaer DJ, Larroche C, Neftel KA. Haemophagocytic syndromes in adults: current concepts and challenges ahead. Swiss Med Wkly. 2005;135:299–314.
20 Balwierz W, Czogała M, Czepko E. Anaplastic large cell lymphoma associated with hemophagocytic lymphohistiocytosis: a case report and review of the literature. Przegl Lek. 2010;67:436–8.
21 Zhu Y, Gao Y, Zhu XH, Chen XY. Diagnosis and treatment of hemophagocytic lymphohistiocytosis in children with HLH-2004 protocol. J Clin Pediatr. 2009;27:718–22.
22 Thompson PA, Allen CE, Horton T, Jones JY, Vinks AA, McClain KL. Severe neurologic side effects in patients being treated for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2009;52:621–5.
23 Escoto H, Ringewald J, Kalpatthi R. Etoposide-related cardiotoxicity in a child with haemophagocytic lymphohistiocytosis. Cardiol Young. 2010;20:105–7.
24 Seo YI, Park R, Choi TY, Shin JW, Won JH, Park HS, et al. A case of therapy-related acute monocytic leukemia following low-dose of etoposide treatment for hemophagocytic lymphohistiocytosis. Korean J Lab Med. 2007;27:244–7.
25 Zhang LJ, Qiu HX, Li JY, Xu J, Wang LL, Hu YX, et al. Clinical analysis of 10 cases of secondary hemophagocytic lymphohistiocytosis treated with HLH-2004 chemotherapy. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2010;18:1525–30.
Funding / potential competing interests: The study was supported in part by the Program for the Development of Innovative Research at the First Affiliated Hospital of NJMU and a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions. We declare that there are no conflicts of interest. We are responsible for the content and drafting of the article.