Creutzfeldt-Jakob disease and mad cows: lessons learnt from yeast cells
DOI: https://doi.org/10.4414/smw.2012.13505
Julia
Hofmann, Hanna
Wolf, Andrea
Grassmann, Verena
Arndt, James
Graham, Ina
Vorberg
Summary
Transmissible spongiform encephalopathies are fatal neurodegenerative diseases that affect mammals including humans. The proteinaceous nature of the infectious agent, the prion, and its propagation, challenge established dogmas in biology. It is now widely accepted that prion diseases are caused by unconventional agents principally composed of a misfolded host-encoded protein, PrP. Surprisingly, major break-throughs in prion research came from studies on functionally unrelated proteins in yeast and filamentous fungi. Aggregates composed of these proteins act as epigenetic elements of inheritance that can propagate their alternative states by a conformational switch into an ordered ß-sheet rich polymer just like mammalian prions. Since their discovery prions of lower eukaryotes have provided invaluable insights into all aspects of prion biogenesis. Importantly, yeast prions provide proof-of-principle that distinct protein conformers can be infectious and can serve as genetic elements that have the capacity to encipher strain specific information. As a powerful and tractable model system, yeast prions will continue to increase our understanding of prion-host cell interaction and potential mechanisms of protein-based epigenetic inheritance.
Abbreviations
BSE: Bovine spongiform encephalopathy
CNS: Central nervous system
PrP: Prion protein
PrPC: Normal cellular isoform of the prion protein
PrPSc: Disease-associated isoform of the prion protein
TSE: Transmissible spongiform encephalopathy
vCJD: Variant Creutzfeldt-Jakob disease
Prion diseases: rare and fatal diseases of mammals
Prion diseases or transmissible spongiform encephalopathies (TSEs) are infectious neurodegenerative diseases that are characterised by proteinaceous deposits and spongiform changes in the central nervous system (CNS) [1]. Astro- and microgliosis, the lack of a lymphocytic inflammatory response and the accumulation of an aggregated host-encoded protein termed prion protein are histopathological features of TSEs. Prion diseases were first identified in the 18th century as a trembling disease in sheep and goats in France and Germany [2] (table 1). Since then, prion diseases have been reported in deer and elk (chronic wasting disease), cattle (bovine spongiform encephalopathy) and other mammals [3]. Human prion diseases can be aetiologically classified into three subsets: sporadic, heritable and acquired or iatrogenic. Sporadic prion disease in humans is known as Creutzfeldt-Jakob disease (CJD) and accounts for 85% of all human TSE cases with an incidence of approximately 1–2 per million of the population [4]. Heritable prion diseases such as fatal familial insomnia and Gerstmann-Sträussler-Scheinker-Syndrome, can be attributed to approximately 30 pathogenic mutations in the prion protein gene [5]. Iatrogenic transmission of CJD has occurred in humans due to the use of TSE-infected surgical instruments, organ transplantation from TSE-donors or through the administration of growth hormones from a TSE-donor [6]. Cases of naturally acquired prion disease include Kuru, a disease of the South Fore tribe of New Guinea, and a new variant of CJD (vCJD) [7, 8].
The outbreak of mad cow disease or bovine spongiform encephalopathy (BSE) in the UK in the mid-1980’s resulted in more than 180,000 cases of BSE with an estimated 1 million cattle infected during the BSE epidemic [9]. In 1989, meat and bone meal was postulated as the source of the infection and was subsequently banned as a food supply for ruminants. In 1994 the first case of human vCJD was reported and it is likely to have been caused by dietary exposure to BSE contaminated beef [10]. To date only 222 cases of vCJD have been reported but since 2004, four cases of vCJD were detected and linked to blood transfusions from pre-clinical TSE-donors raising concerns on the possibility of infection by silent carriers of the TSE agent [11] (table 1).
Protein aggregates as infectious entities
Studies by Griffith in the 1960’s showed that the TSE agent was resistant to all known methods of inactivating nucleic acids, which led to the suggestion that the infectious TSE agent is devoid of a pathogen coding nucleic acid [12]. In 1982 Bolton and colleagues isolated an aggregated and misfolded form of a cellular protein that co-purified with infectivity from scrapie infected hamster brains [13]. The native cellular isoform of the protein was termed PrPC or the prion protein and the isoform associated with infectivity was termed PrPSc. Prusiner postulated that the infectious agent is composed predominantly of the misfolded isoform of the prion protein and is capable of self propagating by converting endogenous PrPC to PrPSc [14]. It is now widely accepted that prions represent protein-based infectious entities that replicate by a process called seeded polymerisation. According to this model, a seed of polymeric fibrillar PrPSc serves as a template for the conformational conversion of PrPC to its pathogenic isoform (fig. 1A). The mechanism of seeded polymerisation is shared by other disease-associated proteins, including A-beta (Alzheimers disease) and huntingtin (Huntington’s Chorea) that form highly ordered ß-sheet rich fibrils termed amyloid [15]. Importantly, self-seeding of ordered protein aggregates is necessary but not sufficient for prion propagation. To increase the number of infectious seeds fibril growth must be followed by fibril fragmentation. Recent data has shown that the most infectious prion particle consists of approximately 14–28 monomers of PrP [16]. The mechanism of prion shearing or fragmentation in mammals is still unknown and no cellular factors have yet been identified to be categorically involved in this process.

Figure 1A
Schematic model of seeded polymerisation, which involves the interaction between a prion seed with PrPC. Partial unfolding of PrPC may promote induction and propagation of PrPC to PrPSc. It is unknown how prion aggregates or fibrils are fragmented into reactive infectious seeds.
The prion protein
The host-encoded PrPC is a 253 amino acid (human; other species vary) protein with two sites for glycosylation, a glycosylphosphatidyl-inositol (GPI) anchor and is translocated to the plasma membrane [17]. PrPC is present in most mammalian cells but is enriched in the cells of the CNS and of the immune system [18]. The exact function of PrPC is unknown, however PrP-knockout mice, with a disrupted PrP gene, show disturbances in circadian rhythm and impaired oxidative stress management [19, 20]. PrP deficient mice also show complete resistance to developing a TSE disease therefore it has been suggested that PrPC plays an important role in prion propagation and TSE pathogenesis [19]. PrPC binds to a diverse range of ligands and might be involved in neuroprotection [21]. A study published in 2010 showed a requirement of PrPC for maintenance of the peripheral myelin sheath [22]. PrPC and PrPSc share the same amino acid sequence but vary in their secondary structure and their biochemical properties [23]. PrPC is a monomer composed mainly of α-helices however during misfolding, PrPC is converted to a β-sheet rich protein, PrPSc, which self propagates and is prone to polymerisation with monomers of itself [24]. The conversion process is believed to occur on the plasma membrane, where PrPC is functionally active, or within the endocytic pathway when PrPC is shuttled away from the plasma membrane for degradation [25–27]. However, the exact mechanism of conversion and the possible involvement of any ancillary co-factors are not fully understood (table 2).
Prion conversion studies in vivo and in vitro have demonstrated that the structural compatibility of PrPC and PrPSc is the major determinant of the species and transmission barriers [28]. The prion species barrier or transmission barrier is the term used to characterise the extended incubation time or ablation of disease, when a species is infected by a TSE-agent originating from another species. A polymorphism at residue 129 (methionine or valine) of the prion protein has been identified to affect susceptibility to sporadic and acquired CJD [29]. So far all confirmed vCJD patients and most cases of sporadic CJD are confined to homozygotes for methionine at this residue. Heterozygozity at residue 129 appears to have a protective effect by inhibiting homologous protein-protein interactions [29].
Conformation of PrPSc might be the basis for different TSE strains
The existence of different prion strains that can be propagated in the same species has long challenged the protein-only hypothesis. Different TSE strains are characterised by variations in neuropathology, and incubation time before clinical symptoms. Since strains can be propagated in the same inbred mouse lines, strain characteristics cannot be encoded by the PrP amino acid sequence. Furthermore, no strain-specific posttranslational modifications of PrPSc have been identified. Consequently, if prions are composed of misfolded prion protein, then different prion strains must arise from conformationally distinct amyloid forms capable of stably self-templating their specific conformations (fig. 1B). Indeed, conformationally distinct isoforms of PrPSc are reproducibly propagated upon serial passage in inbred animal lines [30, 31]. Moreover, biochemical characterisation of different prion strains demonstrate remarkable differences in the biochemical properties of PrPSc molecules, suggesting that strain specific information might indeed be “enciphered” within a specific fold of the core PrPSc aggregate [32].
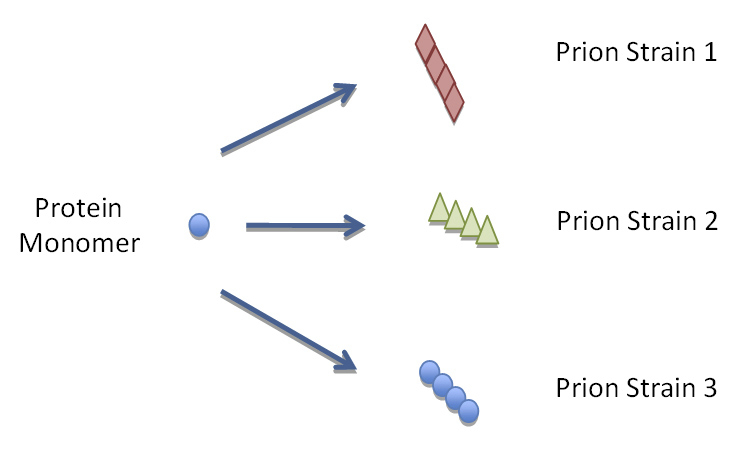
Figure 1B
Yeast and mammalian proteins with prion domains have the ability to misfold into a multitude of different conformations that encipher different biochemical and biophysical properties.
Amplification of prion infectivity in vitro argues against the virion theory
Despite accumulating evidence implicating PrP as the major component of the infectious agent, the protein-only hypothesis is keenly debated by scientists who believe that the infectious agent has a pathogen coding nucleic acid [33]. However, recently designed in vitro assays such as the protein misfolding cyclic amplification (PMCA) have demonstrated that the infectious agent responsible for TSE disease could not be a virus or virion [34]. The PMCA is a procedure that is conceptually analogous to the polymerase chain reaction, where PrPSc content and prion infectivity can be drastically increased by serial cycles of polymer growth and fibril fragmentation. Minute quantities of PrPSc present in brain homogenate or blood from prion-infected animals can be used as a seed to template the conversion of substrate PrPC [34, 35]. Sonication is used in cycles to break up aggregates, thus increasing the number of catalytic sites available for conversion [36]. This assay recapitulates key features of TSE agent replication such as the species barrier and strain characteristics, which are preserved after serial prion amplification in the PMCA. Conventional PMCA uses crude brain homogenates, therefore it cannot be ruled out that there are additional factors present in brain homogenate crucial to the conversion process. Incorporation of additional components such as poly(A)RNA and lipids into the PMCA reaction increased PrPSc and infectivity, thus indicating the possible involvement of these factors in the conversion process [37], although evidence for direct involvement of RNA in in vivo prion formation has not been shown. Alternatively, sulfated glycosaminoglycans may also serve as cellular factors necessary for in vivo and in vitro PrPSc formation [38]. Persuasive evidence that the infectious TSE agent is derived of misfolded PrP was demonstrated when recombinant PrP was converted into a ß-sheet isoform that resulted in a TSE-like disease when injected into animals [39–41]. The fact that these induced prion diseases drastically differed in their clinical and pathological features from known natural prions, make it unlikely that clinical presentation in these animals was due to experimental contamination. Instead, the distinct prion phenotypes observed in vivo were likely the result of different amyloid conformers produced in vitro. However, it has been theorised that the extended incubation times of synthetic prions suggest that only a minor subfraction of PrPSc particles are infectious. Alternatively, synthetic prions are primitive surrogates of PrPSc that might evolve into infectious PrPSc or indeed may serve as a protein scaffold for de novo PrPSc formation in vivo [42].
Prions as epigenetic elements of inheritance
In 1994 the prion hypothesis was extended to include other proteins identified in lower eukaryotes that behave like prions [43] (table 1). The existence of non-Mendelian genetic elements in bakers yeast (Saccharomyces cerevisiae) had long puzzled geneticists. These factors produced phenotypic traits that failed to segregate at mitosis suggesting that their determinants had an extrachromosomal location [43, 44]. Wickner and later Lindquist, provided evidence that these non-Mendelian heritable traits in yeast were caused by self-propagating misfolded isoforms of normal cellular proteins that when in their aggregated state show a loss-of-function phenotype [43, 45]. Similar to mammalian prions, fungal prion proteins can adopt at least two structurally distinct states (monomeric and ordered aggregates) that differ structurally and functionally. Once misfolded, fungal prion proteins aggregate and propagate their abnormal state by templating the conformation onto the natively folded soluble monomeric isoform. Although toxic yeast prion variants might exist, yeast prions are not generally considered to be toxic to yeast and rather constitute epigenetic elements of inheritance that may play an evolutionary role in environmental adaptation [46, 47]. At least eight prions of lower eukaryoteshave been identified (table 1) of which the translation termination factor Sup35 is the most studied [48]. Sup35 promotes ribosomal polypeptide chain release at the stop codon and oligomerisation of the Sup35 prion domain results in non-functional protein complexes that cause a non-sense suppression phenotype [49]. Consequently the misfolded isoform of Sup35 results in a loss of function phenotype characterised by a change in yeast metabolism. Spontaneous conversion of the Sup35 protein into its aggregated prion-like state is a very rare event and occurs at a frequency of 10–5–10–7per cell [49]. Prion-like inheritance has also been demonstrated with the heterokaryon incompatibility factor in the filamentous fungus Podospora anserina [50]. Remarkably fungal prion proteins share little sequence similarity, however the prion domains of several yeast prions contain stretches rich in asparagine (Asn) and glutamine (Gln) residues that are critical for prion formation and propagation. Furthermore, randomisation of prion domains in yeast prions showed that the self-propagating capacity was retained and therefore is related to the amino acid composition rather than a specific sequence [51]. Unique prion variants in yeast, similar to that of mammalian prion strains, have been identified to be associated with specific and distinct metabolic phenotypes [52]. These heritable genetic elements may differ in their biochemical and biophysical properties but they also influence yeast-mating behaviour and differ in their transmission to daughter cells [48, 53].

Table 1
Timeline of important discoveries in mammalian and yeast prion research.
|
Table 2: Comparison of the main characteristics of mammalian and fungal prions. |
| |
TSE agents
|
Fungal prions
|
|
Implicated protein
|
Prion Protein (PrP) |
Sup35, Ure2, Rnq1, Het-s, Cyc8, Swi1, Mot3, Pma1, Sfp1 |
|
Cellular localisation of native isoform
|
GPI-anchored on plasma membrane |
Cytosol |
|
Phenotype associated with prion conformation
|
Fatal transmissible spongiform encephalopathy |
Metabolic |
|
Predicted site of prion formation/propagation
|
Plasma membrane and/or endocytic pathway |
Cytosol |
|
Prion domain of protein
|
Not well defined
(aa 90-231) |
Sup35
(N domain) |
|
Sequence similarity
– Repeat region
– Q and/or N-rich
|
Yes
No |
In Sup35
Yes, except for Het-s, Pma1 |
|
Aggregate fragmentation factors
|
None defined |
Chaperone Hsp104
in conjunction with Hsp70/Hsp40 |
|
Infectivity
|
Yes
Cell to cell, intraspecies and
interspecies
(transmission barrier) |
Yes
cytoduction (cytoplasmic mixing) |
|
Mitotic stability
|
Yes (in cell culture) |
Yes |
|
Transmission barrier
|
Yes |
Yes |
|
Strains
|
Yes |
Yes |
|
Protein-only evidence
|
Supporting evidence |
Yes |
| aa: amino acid |
Yeast prions recapitulate key characteristics of mammalian prions
Since the utilisation of yeast as a model system, our understanding of prion biogenesis has been greatly enhanced. Seminal studies in Saccharomyces cerevisiae demonstrated first proof of principle of the protein-only hypothesis when prion infectivity could be generated in vitro from pure recombinant protein [53–55]. Introduction of fibrillar recombinant fungal proteinin vitro is sufficient to induce a prion phenotype in yeast. This observation has formed the basis for the argument that amyloid fibrils are the molecular basis of yeast prion infectivity. Furthermore, transmission experiments between Saccharomyces cerevisiae and Candida albicans demonstrated a prion transmission barrier when infected with fibrils of the recombinant Sup35 prion domain [54, 56]. The reduction in infectivity in Candida albicans was attributed to the transmission barrier between two different species, a characteristic shared with mammalian prions. Another characteristic of mammalian prions is the existence of unique strains that elicit phenotypically different clinical characteristics. Studies in yeast revealed that distinct fibril types produced from recombinant yeast prion domains induced stable and phenotypically different states in yeast arguing that prion strain variation is decided by the conformation of the prion protein aggregate [53, 57]. Thus, by misfolding into multiple distinct infectious conformations, yeast prions can act as infectious proteins with heritable properties (fig. 1B).
Heterogeneous protein aggregates can serve as imperfect templates for prion induction
The mechanisms of prion misfolding and aggregation in spontaneous forms of TSEs remain elusive and it is the identification of factors that can influence or cross-seed the prion protein conformation that is of great scientific interest. In a study by Derkatch and colleagues to identify factors that affect the induction and propagation of prions in yeast, another cellular prion composed of the host protein Rnq1, was discovered. Rnq1 prions themselves are epigenetic elements that are required for the de novo formation of the Sup35 prion in yeast [58] (fig. 1C). In addition, an unbiased functional screen has identified eleven other Gln/Asn-rich proteins, which when over-expressed, can function as prion-inducing factors. The list of proteins that can promote the de novo conversion of Sup35 to its prion conformation can now potentially be extended to other non-prion proteins. This was directly shown when aggregates of the poly-glutamine rich region encoded by exon I of huntingtin acted as prion-inducing factors [60]. Interestingly, non-polyglutamine containing amyloidogenic proteins such as transthyretin, α-synuclein and synphilin cannot induce aggregation, arguing for the necessity of Gln/Asn-rich domains for prion induction and propagation [60]. If the conformational change of a protein to a prion state can be templated by other prions, or by the over-expression of proteins with Gln/Asn-rich domains, then heterogeneous protein aggregates could catalyse the seeding of the prion conformation [61].
Involvement of the chaperone system in yeast prion replication
Prion replication and propagation crucially depend on fibril elongation and segregation, mechanisms that are not fully understood for mammalian prions. Genetic studies have shown that a chaperone network stringently controls prion propagation in yeast [62]. The central key player of this network is the protein disaggregase Hsp104 for which no mammalian homologue has been identified [63]. Hsp104 is a chaperone that cooperates with the Hsp70 machinery to regulate prion biogenesis for several yeast prions [63]. At moderate expression levels, Hsp104 assists prion propagation by fragmenting prion aggregates and thereby generating new conversion sites, whereas at high concentrations, Hsp104 disassembles fibrils into non-infectious monomers. The activity of Hsp104 is stringently controlled by molecular chaperones of the Hsp70 family and their regulating co-chaperones (fig. 1C) [62]. Other chaperone systems might also be involved in the regulation of prion biogenesis. For example, over-expression of Hsp90 significantly decreases prion induction. The involvement of chaperones in amyloid fragmentation led to the postulation that host cell factors convert an amyloid protein into an infectious aggregate [64]. The association between host cell and aggregates is likely to also influence the fate of amyloidogenic proteins in mammals in general.

Figure 1C
The induction, propagation and fragmentation of mammalian and yeast prions are compared. Partial unfolding of proteins with prion domains and/or the presence of other cellular factors may influence susceptibility to misfolding. (i) In the presence of a prion seed, PrPC is converted to PrPSc thereby elongating the PrPSc fibril. Conversion is likely to be aided by the presence of lipids and/or other co-factors and aggregates are fragmented by an as yet unidentified fragmentation process. (ii) Induction of yeast prions depends on interaction with actin cytoskeleton and/or cross-seeding by other yeast prions or yeast proteins with Gln/Asn-rich domains. Fragmentation of yeast prion aggregates is accomplished by the yeast protein chaperones Hsp104 and Hsp70/ Hsp40.
Identification of other factors affecting yeast prion biogenesis
There is accumulating evidence from genetic studies that prion biogenesis is modulated by a cellular protein degradation system termed the ubiquitin/proteasome system and deficiencies in this system have been shown to increase induction of some yeast prions [65, 66]. Due to the absence of data indicating a direct ubiquitination of yeast prions prior to degradation, it is believed that the ubiquitin/proteasome system instead influences prion biogenesis by regulating ancillary factors. A study by Chernova and colleagues described the regulation of prion biogenesis by ubiquitination and proteasomal degradation of the prion-inducing factor, Lsb2 [67]. Inhibition of its ubiquitination and subsequent degradation resulted in constantly elevated levels of this factor, which in turn increased levels of prion induction. A number of recent studies have been published describing the association between prion biogenesis and the actin cytoskeleton. The yeast prion Sup35 interacts with several proteins of the actin cytoskeleton and alterations of actin polymerisation decrease Sup35 prion induction [68]. In addition, the association of the previously described prion-inducing factor, Lsb2, with the actin cytoskeleton, is required for its prion-inducing function [67]. A different experimental approach was by proteomic analysis, which identified several proteins that associated with yeast prions and may function as yeast prion modifiers [69]. In conclusion, studies on yeast prions have shown a direct regulatory process by the host cell on prion induction and propagation. Therefore, the identification of novel factors that mediate spontaneous induction and propagation of prions in yeast will likely also facilitate the discovery of therapeutic interventions in TSE diseases.
Functional amyloid and beneficial prions
Historically, amyloid has been defined as β-sheet rich extracellular proteinaceous deposits associated with neurodegenerative disorders. Today, amyloid is often defined biophysically as fibrillar ordered protein-aggregates exhibiting characteristic cross-β-sheet x-ray diffraction patterns. Our view of amyloid as disease-associated protein aggregates has drastically changed to incorporate the findings that there are amyloid fibrils performing beneficial functions in prokaryotes as well as lower eukaryotes and even mammals [70, 71]. Cellular processes appear to tightly control functional amyloid formation to prevent uncontrolled spreading and subsequent cell damage. Amyloid has been shown to be an abundant component of natural biofilms formed by a wide range of bacteria. Both silkmoth chorion proteins and spider silk are composed of amyloid fibrils [72, 73]. Other functional amyloid has been proposed in important biological processes; the aggregation of the protein CPEB to amyloid may contribute to the basis for long-term memory in sensory neurons of Aplysia[74]. In mammals, the Pmel17 protein in its amyloid state is a functional component of melanosomes [75]. In addition, it has been shown that even certain hormone peptides form amyloid-like aggregates when packaged into secretory granules [76]. In this case, the obvious advantage for aggregation is that the amyloid conformation allows for a denser packaging of the peptide hormone for future demand. Clearly, these studies have already uncovered that amyloid is much more abundant than previously anticipated.
The identification of the heterokaryon incompatibility factor Het-s in Podospora anserina with gain-of function associated with its amyloid-like state goes beyond the concept of physiological amyloid, showing that some amyloids can even rapidly spread and propagate their aggregated states in a prion-like manner [77]. This has raised the question, how common is the prion phenomenon and could there be more beneficial prions? Proteins with prion functionality are continuously being characterised in yeast and genome wide searches in lower and higher eukaryotes, including mammals, have identified many proteins that may have the potential to act like prions [78]. Clearly, cellular mechanisms exist in the mammalian cytosol that supports prion-like inheritance [79]. Thus, it is tempting to speculate that additional prions will also be discovered in mammals.
Concluding remarks
Over the past few years important work in the model organism,Saccharomyces cerevisiae, has shed new light on the molecular mechanisms that govern prion biogenesis. Research on yeast prions provided first evidence that protein polymers can act as infectious entities and can faithfully store and propagate strain information. This work also illustrated the crucial role of host cell factors for spontaneous prion induction and fragmentation. Whilst these factors are unlikely to be identical in the biogenesis of the TSE agent in mammals (primarily because PrP conversion in mammals is either occurring extracellularly on the plasma membrane or within endocytic vesicles), they highlight that the intimate interaction of amyloidogenic protein aggregates and the host cell is a critical determinant in the adaptation from amyloid to an infectious protein entity. These data become even more valuable as recent studies suggested that several amyloids associated with some of the most prevalent human neurodegenerative diseases might spread in a prion-like manner [80–82]. Therefore, the identification of prion regulatory factors in yeast could facilitate in the characterisation of common mechanisms that underlie the formation and propagation of disease-related amyloid. Studies on prions and amyloid in lower eukaryotes have also brought up the intriguing concept that conversion to the amyloid or prion conformation might not necessarily be detrimental to the host. With the benefit of a tractable model in yeast, it will be the challenge of future research to identify these proteins and to understand the physiological relevance of their amyloid-like conformations in health and disease.
Acknowledgements:We thank Sybille Krauss and Walker Jackson for critical reading of the manuscript.
Authors’ contributions: * These authors contributed equally to the work
References
1 Aguzzi A. Prion diseases of humans and farm animals: Epidemiology, genetics, and pathogenesis. J Neurochem. 2006;97(6):1726–39.
2 McGowan J. Scrapie in sheep. Scott J Agric. 1922;5:365–75.
3 Sigurdson CJ, Miller MW. Other animal prion diseases. Br Med Bull. 2003;66:199–212.
4 Wadsworth JD, Collinge J. Molecular pathology of human prion disease. Acta Neuropathol. 2011;121(1):69–77.
5 Mead S. Prion disease genetics. Eur J Hum Genet. 2006;14(3):273–81.
6 Norrby E. Prions and protein-folding diseases. J Intern Med. 2011;270(1):1–14.
7 Alpers MP. Review. The epidemiology of kuru: Monitoring the epidemic from its peak to its end. Philos Trans R Soc Lond B Biol Sci. 2008;363(1510):3707–13.
8 Collinge J. Variant creutzfeldt-jakob disease. Lancet. 1999;354(9175):317–23.
9 Anderson RM, Donnelly CA, Ferguson NM, Woolhouse ME, Watt CJ, Udy HJ, et al. Transmission dynamics and epidemiology of bse in british cattle. Nature. 1996;382(6594):779–88.
10 Hill AF, Desbruslais M, Joiner S, Sidle KC, Gowland I, Collinge J, et al. The same prion strain causes vcjd and bse. Nature. 1997;389(6650):448–50, 526.
11 Peden AH, Head MW, Ritchie DL, Bell JE, Ironside JW. Preclinical vcjd after blood transfusion in a prnp codon 129 heterozygous patient. Lancet. 2004;364(9433):527–9.
12 Griffith JS. Self-replication and scrapie. Nature. 1967;215(5105):1043–4.
13 Bolton DC, McKinley MP, Prusiner SB. Identification of a protein that purifies with the scrapie prion. Science. 1982;218(4579):1309–11.
14 Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216(4542):136–44.
15 Goedert M, Clavaguera F, Tolnay M. The propagation of prion-like protein inclusions in neurodegenerative diseases. Trends Neurosci. 2010;33(7):317–25.
16 Silveira JR, Raymond GJ, Hughson AG, Race RE, Sim VL, Hayes SF, et al. The most infectious prion protein particles. Nature. 2005;437(7056):257–61.
17 Stahl N, Borchelt DR, Hsiao K, Prusiner SB. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell. 1987;51(2):229–40.
18 Linden R, Martins VR, Prado MA, Cammarota M, Izquierdo I, Brentani RR. Physiology of the prion protein. Physiol. Review. 2088;88:673–728.
19 Bueler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, et al. Mice devoid of prp are resistant to scrapie. Cell. 1993;73(7):1339–47.
20 Wong BS, Liu T, Li R, Pan T, Petersen RB, Smith MA, et al. Increased levels of oxidative stress markers detected in the brains of mice devoid of prion protein. J Neurochem. 2001;76(2):565–72.
21 Linden R, Martins VR, Prado MA, Cammarota M, Izquierdo I, Brentani RR. Physiology of the prion protein. Physiol Rev. 2008;88(2):673–728.
22 Bremer J, Baumann F, Tiberi C, Wessig C, Fischer H, Schwarz P, et al. Axonal prion protein is required for peripheral myelin maintenance. Nat Neurosci. 2010;13(3):310–8.
23 Stahl N, Baldwin MA, Teplow DB, Hood L, Gibson BW, Burlingame AL, et al. Structural studies of the scrapie prion protein using mass spectrometry and amino acid sequencing. Biochemistry. 1993;32(8):1991–2002.
24 Gasset M, Baldwin MA, Fletterick RJ, Prusiner SB. Perturbation of the secondary structure of the scrapie prion protein under conditions that alter infectivity. Proc Natl Acad Sci U S A. 1993;90(1):1–5.
25 Borchelt DR, Taraboulos A, Prusiner SB. Evidence for synthesis of scrapie prion proteins in the endocytic pathway. J Biol Chem. 1992;267(23):16188–99.
26 Taraboulos A, Scott M, Semenov A, Avrahami D, Laszlo L, Prusiner SB. Cholesterol depletion and modification of cooh-terminal targeting sequence of the prion protein inhibit formation of the scrapie isoform. J Cell Biol. 1995;129(1):121–32.
27 Marijanovic Z, Caputo A, Campana V, Zurzolo C. Identification of an intracellular site of prion conversion. PLoS Pathog. 2009;5(5):e1000426
28 Horiuchi M, Priola SA, Chabry J, Caughey B. Interactions between heterologous forms of prion protein: Binding, inhibition of conversion, and species barriers. Proc Natl Acad Sci U S A. 2000;97(11):5836–41.
29 Wadsworth JD, Asante EA, Desbruslais M, Linehan JM, Joiner S, Gowland I, et al. Human prion protein with valine 129 prevents expression of variant cjd phenotype. Science. 2004;306(5702):1793–6.
30 Telling GC, Parchi P, DeArmond SJ, Cortelli P, Montagna P, Gabizon R, et al. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science. 1996;274(5295):2079–82.
31 Safar J, Wille H, Itri V, Groth D, Serban H, Torchia M, et al. Eight prion strains have prp(sc) molecules with different conformations. Nat Med. 1998;4(10):1157–65.
32 Bessen RA, Kocisko DA, Raymond GJ, Nandan S, Lansbury PT, Caughey B. Non-genetic propagation of strain-specific properties of scrapie prion protein. Nature. 1995;375(6533):698–700.
33 Manuelidis L, Yu ZX, Barquero N, Mullins B. Cells infected with scrapie and creutzfeldt-jakob disease agents produce intracellular 25-nm virus-like particles. Proc Natl Acad Sci U S A. 2007;104(6):1965–70.
34 Saborio GP, Permanne B, Soto C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature. 2001;411(6839):810–3.
35 Castilla J, Saa P, Hetz C, Soto C. In vitro generation of infectious scrapie prions. Cell. 2005;121(2):195–206.
36 Soto C, Saborio GP, Anderes L. Cyclic amplification of protein misfolding: Application to prion-related disorders and beyond. Trends Neurosci. 2002;25(8):390–4.
37 Deleault NR, Geoghegan JC, Nishina K, Kascsak R, Williamson RA, Supattapone S. Protease-resistant prion protein amplification reconstituted with partially purified substrates and synthetic polyanions. J Biol Chem. 2005;280(29):26873–9.
38 Wong C, Xiong LW, Horiuchi M, Raymond L, Wehrly K, Chesebro B, et al. Sulfated glycans and elevated temperature stimulate prp(sc)-dependent cell-free formation of protease-resistant prion protein. EMBO J. 2001;20(3):377–86.
39 Legname G, Baskakov IV, Nguyen HO, Riesner D, Cohen FE, DeArmond SJ, et al. Synthetic mammalian prions. Science. 2004;305(5684):673–6.
40 Makarava N, Kovacs GG, Bocharova O, Savtchenko R, Alexeeva I, Budka H, et al. Recombinant prion protein induces a new transmissible prion disease in wild-type animals. Acta Neuropathol. 2010;119(2):177–87.
41 Wang F, Wang X, Yuan CG, Ma J. Generating a prion with bacterially expressed recombinant prion protein. Science. 2010;327(5969):1132–5.
42 Baskakov IV. The reconstitution of mammalian prion infectivity de novo. FEBS J. 2007;274(3):576–87.
43 Wickner RB. [ure3] as an altered ure2 protein: Evidence for a prion analog in saccharomyces cerevisiae. Science. 1994;264(5158):566–9.
44 Aigle M, Lacroute F. Genetical aspects of [ure3], a non-mitochondrial, cytoplasmically inherited mutation in yeast. Mol Gen Genet. 1975;136(4):327–35.
45 Patino MM, Liu JJ, Glover JR, Lindquist S. Support for the prion hypothesis for inheritance of a phenotypic trait in yeast. Science. 1996;273(5275):622–6.
46 Halfmann R, Lindquist S. Epigenetics in the extreme: Prions and the inheritance of environmentally acquired traits. Science. 2010;330(6004):629–32.
47 Wickner RB, Edskes HK, Bateman D, Kelly AC, Gorkovskiy A. The yeast prions [psi+] and [ure3] are molecular degenerative diseases. Prion. 2011;5(4):
48 Wickner RB, Edskes HK, Shewmaker F, Nakayashiki T, Engel A, McCann L, et al. Yeast prions: Evolution of the prion concept. Prion. 2007;1(2):94–100.
49 Uptain SM, Lindquist S. Prions as protein-based genetic elements. Annu Rev Microbiol. 2002;56:703–41.
50 Coustou V, Deleu C, Saupe S, Begueret J. The protein product of the het-s heterokaryon incompatibility gene of the fungus podospora anserina behaves as a prion analog. Proc Natl Acad Sci U S A. 1997;94(18):9773–8.
51 Ross ED, Edskes HK, Terry MJ, Wickner RB. Primary sequence independence for prion formation. Proc Natl Acad Sci U S A. 2005;102(36):12825–30.
52 Schlumpberger M, Prusiner SB, Herskowitz I. Induction of distinct [ure3] yeast prion strains. Mol Cell Biol. 2001;21(20):7035–46.
53 Tanaka M, Chien P, Naber N, Cooke R, Weissman JS. Conformational variations in an infectious protein determine prion strain differences. Nature. 2004;428(6980):323–8.
54 Sparrer HE, Santoso A, Szoka FC, Jr., Weissman JS. Evidence for the prion hypothesis: Induction of the yeast [psi+] factor by in vitro- converted sup35 protein. Science. 2000;289(5479):595–9.
55 Brachmann A, Baxa U, Wickner RB. Prion generation in vitro: Amyloid of ure2p is infectious. EMBO J. 2005;24(17):3082–92.
56 Chien P, Weissman JS, DePace AH. Emerging principles of conformation-based prion inheritance. Annu Rev Biochem. 2004;73:617–56.
57 King CY, Diaz-Avalos R. Protein-only transmission of three yeast prion strains. Nature. 2004;428(6980):319–23.
58 Derkatch IL, Bradley ME, Zhou P, Chernoff YO, Liebman SW. Genetic and environmental factors affecting the de novo appearance of the [psi+] prion in saccharomyces cerevisiae. Genetics. 1997;147(2):507–19.
59 Scherzinger E, Sittler A, Schweiger K, Heiser V, Lurz R, Hasenbank R, et al. Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrils: Implications for huntington's disease pathology. Proc Natl Acad Sci U S A. 1999;96(8):4604–9.
60 Derkatch IL, Uptain SM, Outeiro TF, Krishnan R, Lindquist SL, Liebman SW. Effects of q/n-rich, polyq, and non-polyq amyloids on the de novo formation of the [psi+] prion in yeast and aggregation of sup35 in vitro. Proc Natl Acad Sci U S A. 2004;101(35):12934–9.
61 Derkatch IL, Bradley ME, Hong JY, Liebman SW. Prions affect the appearance of other prions: The story of [pin(+)]. Cell. 2001;106(2):171–82.
62 Sweeny EA, Shorter J. Prion proteostasis: Hsp104 meets its supporting cast. Prion. 2008;2(4):135–40.
63 Shorter J, Lindquist S. Hsp104, hsp70 and hsp40 interplay regulates formation, growth and elimination of sup35 prions. EMBO J. 2008;27(20):2712–24.
64 Wegrzyn RD, Bapat K, Newnam GP, Zink AD, Chernoff YQ. Mechanism of prion loss after Hsp104 inactivation in yeast. Mol Cell Biol. 2001;14:4656–69.
65 Allen KD, Chernova TA, Tennant EP, Wilkinson KD, Chernoff YO. Effects of ubiquitin system alterations on the formation and loss of a yeast prion. J Biol Chem. 2007;282(5):3004–13.
66 Tyedmers J, Madariaga ML, Lindquist S. Prion switching in response to environmental stress. PLoS Biol. 2008;6(11):e294
67 Chernova TA, Romanyuk AV, Karpova TS, Shanks JR, Ali M, Moffatt N, et al. Prion induction by the short-lived, stress-induced protein lsb2 is regulated by ubiquitination and association with the actin cytoskeleton. Mol Cell. 2011;43(2):242–52.
68 Ganusova EE, Ozolins LN, Bhagat S, Newnam GP, Wegrzyn RD, Sherman MY, et al. Modulation of prion formation, aggregation, and toxicity by the actin cytoskeleton in yeast. Mol Cell Biol. 2006;26(2):617–29.
69 Redeker V, Hughes C, Savistchenko J, Vissers JP, Melki R. Qualitative and quantitative multiplexed proteomic analysis of complex yeast protein fractions that modulate the assembly of the yeast prion sup35p. PLoS One. 2011;6(9):e23659.
70 Fowler DM, Koulov AV, Balch WE, Kelly JW. Functional amyloid – from bacteria to humans. Trends Biochem Sci. 2007;32(5):217–24.
71 Badtke MP, Hammer ND, Chapman MR. Functional amyloids signal their arrival. Sci Signal. 2009;2(80):pe43.
72 Iconomidou VA, Chryssikos GD, Gionis V, Galanis AS, Cordopatis P, Hoenger A, et al. Amyloid fibril formation propensity is inherent into the hexapeptide tandemly repeating sequence of the central domain of silkmoth chorion proteins of the a-family. J Struct Biol. 2006;156(3):480–8.
73 Kenney JM, Knight D, Wise MJ, Vollrath F. Amyloidogenic nature of spider silk. Eur J Biochem. 2002;269(16):4159–63.
74 Si K, Choi YB, White-Grindley E, Majumdar A, Kandel ER. Aplysia cpeb can form prion-like multimers in sensory neurons that contribute to long-term facilitation. Cell. 2010;140(3):421–35.
75 Fowler DM, Koulov AV, Alory-Jost C, Marks MS, Balch WE, Kelly JW. Functional amyloid formation within mammalian tissue. PLoS Biol. 2006;4(1):e6.
76 Maji SK, Perrin MH, Sawaya MR, Jessberger S, Vadodaria K, Rissman RA, et al. Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science. 2009;325(5938):328–32.
77 Maddelein ML, Dos Reis S, Duvezin-Caubet S, Coulary-Salin B, Saupe SJ. Amyloid aggregates of the het-s prion protein are infectious. Proc Natl Acad Sci U S A. 2002;99(11):7402–7.
78 Alberti S, Halfmann R, King O, Kapila A, Lindquist S. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell. 2009;137(1):146–58.
79 Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ. Mavs forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell. 2011;146(3):448–61.
80 Aguzzi A. Cell biology: Beyond the prion principle. Nature. 2009;459(7249):924–5.
81 Krammer C, Kryndushkin D, Suhre MH, Kremmer E, Hofmann A, Pfeifer A, et al. The yeast sup35nm domain propagates as a prion in mammalian cells. Proc Natl Acad Sci U S A. 2009;106(2):462–7.
82 Aguzzi A, Rajendran L. The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron. 2009;64(6):783–90.