Artificial muscle: facts and fiction
DOI: https://doi.org/10.4414/smw.2011.13319
Summary
Mechanical devices are sought to support insufficient or paralysed striated muscles including the failing heart. Nickel-titanium alloys (nitinol) present the following two properties: (i) super-elasticity, and (ii) the potential to assume different crystal structures depending on temperature and/or stress. Starting from the martensite state nitinol is able to resume the austenite form (state of low potential energy and high entropy) even against an external resistance. This one-way shape change is deployed in self-expanding vascular stents. Heating induces the force generating transformation from martensite to the austenite state while cooling induces relaxation back to the martensite state. This two-way shape change oscillating between the two states may be used in cyclically contracting support devices of silicon-coated nitinol wires. Such a contractile device sutured to the right atrium has been tested in vitro in a bench model and in vivo in sheep. The contraction properties of natural muscles, specifically of the myocardium, and the tight correlation with ATP production by oxidative phosphorylation in the mitochondria is briefly outlined. Force development by the nitinol device cannot be smoothly regulated as in natural muscle. Its mechanical impact is forced onto the natural muscle regardless of the actual condition with regard to metabolism and Ca2+-homeostasis. The development of artificial muscle on the basis of nitinol wires is still in its infancy. The nitinol artificial muscle will have to prove its viability in the various clinical settings.
Scope of the subject
In a review in this issue of Swiss Medical Weekly [1] Piergiorgio Tozzi discusses functions and applications of new technologies that enable the construction of “artificial muscle”. These devices, dubbed “smart materials”, apparently may “behave” like biological systems by “converting electrical energy into movement and contraction”. Contraction and expansion operates silently like “natural muscles”. The author emphasises a variety of potential medical applications where these devices may restore or replace the mechanical function of failing muscle including wrapping around the heart for improvement of systolic contractility or placing the new constructs on the atrium to support the blood ejection into the ventricle during chronic atrial fibrillation. Further potential applications may include artificial sphincters to combat urinary and fecal incontinence, restoration of eye blinking in patients with facial paralysis due to stroke or nerve injury, reconstruction of peristaltic esophagus and esophageal sphincter movement. These are all clinically relevant ailments deserving intensive research for ingenious resolutions. The purpose of this editorial is to complement the review on artificial muscle [1] by focusing on the questions what is similar and what is dissimilar between natural muscle and the new constructs, what are the biological consequences and potential dangers resulting from the differences between them. First let's consider the normal function of skeletal and heart muscle.
Contraction of natural muscle
The salient feature of natural muscle consists in its highly structured arrangement [2] of the actin filament tracks and the interdigitating bipolar myosin filaments in the middle of the sarcomere (fig. 1). The motor domains of myosin interact repeatedly with actin in an irregular manner thereby pulling the actin filaments step-by-step toward the sarcomere center. The irregular (stochastic) interaction of myosin crossbridges with actin allows for smooth sliding of the filaments past each other and ensures continuity of force generation. This cyclic actin interaction is tightly coupled to ATP splitting which provides the energy for contraction. Binding of ATP to myosin heads and the subsequent release of the products ADP and inorganic phosphate induces a sequence of protein conformational changes that governs the interaction of the crossbridges with actin [3]. First, the myosin heads homing into the actin binding sites are guided by long-range electro-static forces while, second, the strong interaction that serves as starting point for the power stroke, is primarily governed by hydrophobic protein-protein contacts. After the working cycle has finished, a new ATP binds to myosin and the interaction between the two proteins becomes weak. The crossbridge detaches from actin and hydrolysis of ATP resets the original myosin conformation (recovery stroke) ready to go again. In this way chemical energy is directly converted into directional mechanical energy. Most importantly the length of both actin and myosin filaments does not change during contraction (fig. 1).
In vitro motility assays using a feedback enhanced laser trap system allows direct measurement of force and displacement that results from the interaction of a single myosin molecule with a single suspended actin filament [4]. Discrete stepwise movements averaging ~11 nm were seen under conditions of low load. Single force transients, averaging 3–4 picoNewtons, were measured under isometric conditions. Around 10 billion of myosin crossbridges are required to keep a force of one gram in balance. The maximal isometric force development of striated muscle amounts to around 4 kg per square cm. In vivo, myosin crossbridge stroke distance depends on the resistance the muscle has to work against. This distance tends to zero at isometric contraction (no external work) when the muscle is delivering internal work only, i.e., the myosin crossbridges remain relatively longer attached to resist the external force. Slower moving crossbridges in isometric contraction burn less ATP per force developed (higher efficiency) than fast moving crossbridges in isotonic contraction (fast shortening of unloaded muscle).
The giant protein titin (the largest known human protein) constitutes the stress-responsive elements (third sarcomeric filament system) that protect the sarcomeres from being overstretched and injured during passive extension by antagonist skeletal muscles and during diastolic distension in the heart [5]. One molecule of titin extends from the sarcomere middle at the M-band over the entire half sarcomere (~1 µm) right to the Z-disc. Titin is a modular protein built for 90% of immunoglobulin and fibronectin (FN3)-like domains; the remainder is composed of insertions of unique sequences with different elastic properties. Various isoforms in skeletal and cardiac muscles derive from one single human titin gene. The titin isoforms contain around 27,000 up to 38,000 amino acid residues with molecular masses of 3000–4200 kDa. The titin springs afford the sarcomeres long-range elasticity and determine myocardial passive tension. In addition, titin may coordinate the precise assembly of sarcomeres during muscle development and cardiac hypertrophy.
Functional coupling of contraction with energy production
This biological linear motor system was so successful during evolution that it has been adopted by all kinds of striated muscle (skeletal and heart muscles). A variation of the theme with much longer myosin and actin filaments than in striated muscle, not organised in sarcomere units, is found in smooth muscles. The sarcomere (1.9 up to 2.6 µm in length) presents the smallest contractile unit. In striated muscle the sarcomeres are longitudinally arranged in series from few (in small muscle fibers) up to 10 thousand and more in fibers of large muscles. 10 single myosin crossbridge steps of nearly 10 nm each would suffice to shorten a sarcomere by 10%. Consequently all sarcomeres in series shorten by the same distance and thus the entire muscle would shorten by 10%. 5–15% shortening is indeed the normal range for muscle contraction in vivo. Sarcomeres of similar size and structure are found in the muscles of the Etruscan Shrew (smallest living terrestrial mammal with a body weight of 2 gm and heart weight of 12 mg) as well as in the largest living mammal ever, the Blue Whale (body weight of ~100,000 kg and heart weight of 600 kg) [6]. Likewise the maximal mechanical performance of all sarcomeric muscles ranges between 40 and 200 Watt per kg muscle. In striated muscle the performance efficiency in relation to ATP consumption is around 50% [7].
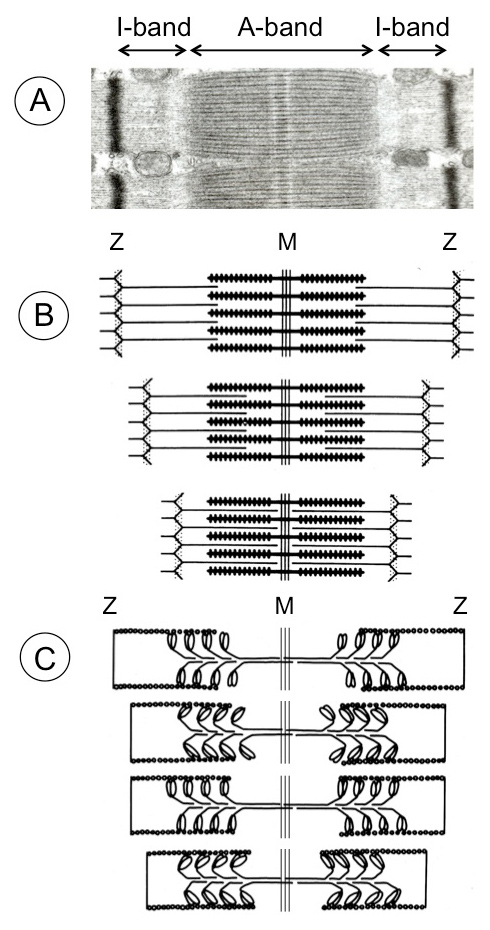
Figure 1
Sarcomere shortening of mammalian skeletal muscle. (A) Electron micrograph of sarcomere displaying A-band (anisotropic) consisting of myosin thick filaments with the interdigitating actin filaments and the M-band proteins in the middle. The I-band (isotropic) contains the thin actin filaments reaching from the Z-discs (black) toward the A-band. The third filamentous structure (not visible in the graph) consists of the giant protein titin. Each titin molecule extends from the sarcomere middle over half the sarcomere length to the Z-disc. Several dozens additional proteins are associated with the three major structural proteins. (B) During muscle contraction the sarcomeres shorten by sliding of the actin filaments toward the sarcomere center while the myosin and actin filaments preserve their original length. (C) The sliding of the two filamentous systems is brought about by repetitive fixation of the myosin heads (crossbridges) at actin, bending toward the sarcomere middle, followed by detachment and fixation again in a new position. This cyclic movement of the myosin heads is coupled to ATP hydrolysis that provides the energy. In reality the myosin heads are not moving in concert like the athletes in a rowing boat (as depicted in (C)) but are moving irregularly (stochastic).
The energy consumption by muscle contraction must, however, be balanced with the energy supply by the cell metabolism. Considering the energy required for restitution of the ATP used for contraction and for the subsequent relaxation processes as well, the overall efficiency drops to about half (~25%). This implies that in a complete contraction-relaxation cycle the metabolic restitution processes operate at a similar efficiency like the chemo-mechanical transduction of the contractile machinery. The ultimate efficiency delivered by the muscle is further diminished to ~10% by internal friction and counterforce of the antagonist muscles. Similarly, also myocardial contraction works with an overall efficiency of around 10% [8]. In skeletal muscle the energy consumption increases 1000-fold when going from rest to isometric contraction [7]. By going from isometric to isotonic contraction (fast contraction under low load), the energy consumption increases a further three times. This drastic increase in energy consumption occurring within a fraction of a second requires fast and precise coordination with the energy producing metabolism. This coordination is ensured by the Ca2+ signal that triggers contraction by Ca2+ binding to troponin-C on the actin filament [9]. At the same time Ca2+ also stimulates the ATP production by activating the phosphorylase kinase (catabolism of muscle glycogen), key enzymes of the tricarboxylic acid cycle in the mitochondrial matrix and the ATP synthase (complex V of the respiratory chain) at the inner mitochondrial membrane [10]. The intimate coupling of mechanical work with metabolism is decisive for cardiac function and particularly traceable in the heart.
Heart failure may be due to myocardial starving
The hypothesis that the failing heart is nutritionally starving has lately been plausibly discussed [11, 12]. The heart amounts to 0.4% of a 70 kg human, yet it consumes ~10% of the total energy expenditure at rest. During physical activity the heart can raise its performance from 10 Watt at rest up to a maximal output of ~100 Watt in an athlete at peak activity. The heart pumps 70 ml blood per beat (stroke volume), ~5 liters per min, 300 liters per hour, 7000 liters per day, 2.5 mill liters per year, and around 210 mill liters during a life time of 80 years (corresponding to a train with 10,000 tank wagons). The brain (1.3 kg) accounts for almost 2% body weight and consumes around 20% of the total energy expenditure [13]. The total brain performance oscillates around 20 Watt during day and night (merely a dim 20 Watt bulb). On a per organ weight basis this is less than half the output of the heart at rest, let alone under 10-times higher load. This illustrates how mechanical work is much more expensive than information processing.
The costs for ATP to support myocardial contraction is under normal conditions met to 70–90% by catabolism of free fatty acids via beta-oxidation, the tricarboxylic acid cycle (Krebs cycle) and oxidative phosphorylation (respiratory chain) in the mitochondria. To bring the energy from the mitochondria to the different energy consuming structures in the cardiomyocyte (primarily contracting myofibrils and ion pumps such as Ca2+-pumps and Na+/K+-pumps), the energy-rich phosphoryl group is transferred by the creatine kinase (CK) from ATP to creatine giving rise to ADP and phosphocreatine. This transphosphorylation occurs in the mitochondrial intermembrane space. The ADP is quickly re-phosphorylated to ATP at the mitochondrial inner membrane. The phosphocreatine is in equilibrium throughout the cytoplasm and its phosphoryl group is readily transferred once more to ADP where energy in the form of ATP is required. CK and creatinephosphate represent an efficient transport shuttle between the mitochondria where ATP is produced and the cellular loci where energy is required [11].
In normal ventricular tissue intracellular concentrations are 8–10 mmol/L for ATP, <50 µmol/L for ADP, <1 mmol/L for inorganic phosphate (Pi), and >200 mmol/L for the phosphorylation potential (PP). The ratio of ATP / ADP • Pi, represents PP that determines the free energy available from hydrolysis of ATP to drive the ATP-requiring reactions [11]. Biochemical tools cannot provide accurate measures of these phosphate compounds. This only became possible with the advent of nuclear magnetic resonance (NMR) spectroscopy and positron emission tomography (PET). The entire cardiac ATP pool just suffices for a few heartbeats. It would be exhausted within one minute at rest and in 10 seconds at high workload [14]. Therefore, ATP needs to be furnished continuously. The heart generates ~30 kg ATP per day (~50 times its own weight). The phosphocreatine pool constitutes the immediate back-up system for ADP re-phosphorylation in the cytoplasm. The total creatine concentration amounts to 30–40 mmol/L of which about two thirds are phosphorylated [15, 16]. This energy-rich phosphocreatine pool is of paramount importance since its phosphotransfer rate to ADP by the CK is ~10-times faster than that for ATP production by oxidative phosphorylation in the mitochondria. In severely failing heart the levels of phosphocreatine and ATP decline by 60–70% and 30–40%, respectively. Under high energy demand additional substrates such as glucose, pyruvate, lactate and ketone bodies are incurred for ATP production (the heart is an omnivore). The relative contributions of the different metabolic pathways for ATP production change in response to changes in fuel supply, hormonal and neural signals; but all these alternative pathways for ATP production are considerably slower than is re-phosphorylation of ADP from phosphocreatine. Of note, the contractility of isolated muscle fibers from failing hearts tested in vitro is usually undistinguishable from those of healthy hearts. Taken together, the unfaltering mechanical activity of the heart throughout the entire life and its tight coupling to metabolism presents a formidable task for a potential artificial muscle. Any physical activity of the heart muscle imprints its characteristics on metabolism and tissue remodeling.
Nitinol as artificial muscle
Although natural muscles are chemically powered with a high-energy-density fuel such as ATP, artificial mechanical elements (actuators) may be driven by piezoelectric, electrostrictive, or electrochemical principles [17]. In addition, chemically powered artificial muscles based on polymer gels have also been demonstrated many years ago. Most of these devices depend on large amounts of energy that renders their application in biology impracticable at present. The properties of newly developed nickel-titanium alloys, also known as nitinol, seem better suited for the construction of “artificial muscles” for medical applications [1]. Its outstanding properties are twofold; one is the so-called super-elasticity or pseudo-elasticity. The elastic deformation of conventional materials such as stainless steel or cobalt-based alloys is limited to about 1% strain whereby elongation increases and decreases linearly (proportionally) with the applied force. Deformation of nitinol by more than 10% strain can be elastically recovered. Nitinol is an intermetallic compound that may assume two kinds of crystal structures: a cubic crystal structure above a certain transition temperature referred to as austenite, and at lower temperature a more complicated monoclinic crystal structure known as martensite [18]. Variation in temperature or application of stress can induce the phase transition between the two. Upon reversion from the martensite to the original austenite structure, regardless of whether the martensite phase was deformed, nitinol assumes its original shape. From this backward phase transition derives the second remarkable property of nitinol dubbed “shape memory”. That has, of course, nothing to do with memory. The austenite has a lower potential energy (higher entropy) than the martensite state. The transition between the two states can be triggered by energy input from the environment such as heat or stress. In other words, the austenite configuration presents the stable equilibrium. The martensite state then presents a local minimum in the potential energy landscape of nitinol that can persist until the local energy barrier is overcome and transition to austenite is triggered. Nitinol may be deformed or compressed at low temperature and subsequently heated to recover its original austenite shape. As an aside, memory applies to a totally different type of quality in the nervous system, namely being able to encode and store incoming new information in variable brain structures. Storage of new information goes along with increased order concomitant with lower entropy.
The oscillation between the austenite and martensite phase can be exploited for technical and medical uses. Depending on the macroscopic structure great forces can be produced by preventing the reversion of deformed martensite to austenite. Early self-expanding nitinol stents were first compressed, percutaneously placed into the vessel and finally heated to the transition temperature for expansion [19]. Today’s nitinol stents are self-expanding without the need for post-deployment heating. This is achieved by manufacturing the nitinol material to respond to a defined temperature range. To avoid premature expansion, the compressed stent is placed in a retractable protective covering during delivery. This transformation is known as “one-way shape memory” effect.
Nitinol wires subject to repetitive heating may concomitantly contract and relax by oscillating between the two physical states, and may thus be used as muscle replacement. However, there are still substantial caveats with continuous use (see below). In the case of repetitive contractions, electrical heating causes the wires to contract by going from a pre-set elongated form (martensite) to the shorter austenite conformation. On cutting off the electrical current, the nitinol wires transform (below the critical transition temperature) back to the original elongated martensite form ready for the next cycle. This is known as “two-way shape memory” effect. However, the working efficiency is rather low around 10%. Internal friction and tissue resistance will further lower the overall efficiency of nitinol artificial muscle considerably.
Nitinol is exceedingly difficult to manufacture because it requires an exceptionally tight compositional control [20]. It is typically composed of 50–51% nickel by atomic percent (55–56% by weight percent). Small changes in the composition can change the transition temperature of the alloy significantly. Titanium is very reactive and may combine with oxygen or carbon. Every reacted titanium atom will be drawn from the nickel-titanium lattice thus shifting the composition and lowering the transformation temperature. Convenient super-elastic temperature ranges are from –20 up to +60 ºC. A combination of heat treatment and cold processing is essential in controlling the properties of nitinol production for different purposes [20].
Known unknowns
In the accompanying review Tozzi [1] discusses a variety of potential medical applications for nitinol devices as adjunct for muscle support or for fully fledged muscle replacement. The experience of the research group in Lausanne extends to atrial assist devices comprising radially arranged, silicone coated, nitinol wires suspended in a plastic ring 40–55 mm in diameter. The plastic ring can be sutured onto the external surface of the right atrium. This mechanical support device has been tested in vitro in bench models as well as in vivo in sheep [21]. In vivo the device was operated by intermittent electric currents of 10 V, 300 mA for 100 ms powered by a battery. The impulse rate was controlled by a pacemaker-like unit that senses the ventricular activity and triggers the current after a delay of 200–500 ms. Measurements were taken during rapid atrial pacing (600 beats per min) of healthy sheep provoking hemodynamics like in atrial fibrillation. Despite the moderate force development, the assist device improved the right atrial ejection fraction (RAEF) from 7% during rapid pacing to 21%. In sinus rhythm without rapid pacing, the baseline RAEF was 41%.
About half of the patients with chronic end-stage heart failure suffer from atrial fibrillation despite previous ablation treatment [22]. Factors responsible for the decrease of cardiac output during atrial fibrillation are loss of synchronous atrial mechanical activity, irregular ventricular response, rapid heart rate, and impaired coronary flow. The atrial nitinol support is thought to restore the coordinated pump function of the fibrillating atria with the ventricles and to reduce the risk of atrial embolism [21].
Several questions regarding the atrial support by nitinol artificial muscle may be asked: (i) what is the transition temperature of the presently used nitinol wires. Nitinol has a poor thermal conductivity. In addition, the wires are embedded in an insulating silicon sheet. Although the highest temperature measured at the right atrial surface was around 40 ºC, the actual temperature of the wires might be considerably higher and the heat is difficult to remove. Can that interfere with the relaxation process that depends on fast cooling below the transition temperature. (ii) Furthermore, where the silicon-coated wires are in direct contact with the atrial surface a great deal of the heat will flow locally through the tissue to be removed by the blood stream. These local thermal fluxes might not show up in temperature measurement of a wider surface area. (iii) Although the nitinol wires of today may allow for billions of transition cycles in vitro what is the potential life time in vivo when these cycles are connected to force delivery. (iv) The heart is beating 35,000,000-times per year. Could the rapid temperature fluctuations affect the wires as well as the silicon coating and limit their lifetime. (v) Electropolished (with a thin titanium oxide surface layer) nitinol stents have excellent corrosion resistance with breakdown potentials greater than 800 mV whereas the breakdown potential of non-electropolished stents ranges around 200 mV. What is the approximate breakdown potential of the employed nitinol wires. (vi) The atrial assist device is equipped with a transcutaneous energy transfer system for recharging the implanted battery. What is the capacity of the battery and how often does it require recharging.
Unknown unknowns
The unknown unknowns are potentially more dangerous than the known unknowns. While the physico-mechanical properties of nitinol alloys can be defined, the biological repercussions by artificial assist devices are unknown, but, nevertheless, most important. An advantage of the nitinol atrial assist device is that it sits as backpack on the outside surface of the atrium and has no direct contact with the bloodstream. Anticoagulation therapy is not required and the danger of infection is drastically reduced as compared to the conventional ventricular assist devices (VAD). Nevertheless, signs of acute (within a couple of days) and chronic (over 4 weeks) inflammation show up where the artificial muscle is in contact with the atrium, though neither tissue necrosis nor apoptosis was observed. Long-term effects have not been tested so far, but it may be expected that the constant friction between the sutured nitinol device and the wall surface during the contraction-relaxation cycles entertain chronic inflammation and fibrosis. While the nitinol device supports the transport of the blood from the atrium to the ventricle, chronic inflammation and fibrosis could be accompanied by progressive remodeling affecting the Ca2+-homeostasis, metabolism, and electrical properties. In patients all three aspects can and should be addressed by appropriate pharmacological treatment in conjunction with the mechanical nitinol device. Only a concerted effort can have a fair chance to combat or at least to delay the inexorably fatal disease of end-stage heart failure.
Another important question pertains to the indication: when should the atrial nitinol device be applied, at which time point in the course of progressing chronic heart failure, or at what severity of the disease. No data are available at present. The nitinol device permits a thoracoscopic minimally invasive implantation. In fact, two such devices could be placed, one on the right and he other on the left atrium [21]. Although the hemodynamic performance of the atrial nitinol device is significantly lower than is the case with current VADs, it could, nevertheless, be used as bridge-to-transplant (the most common application of current VADs) for critically ill patients awaiting heart transplantation. It could also be used as so-called bridge-to-bridge if a long-term VAD with greater surgical risks than with the nitinol device is envisaged after stabilisation or improvement of multiorgan failure. Since the permanent success of sinus rhythm after cardioversion and/or catheter ablation is rather low (40–60% success), implantation of an atrial nitinol device could also be considered to serve as long-term treatment of atrial fibrillation.
Conventional VADs (mostly implanted in the left ventricle (LVAD) and in 20–30% of patients also a second device in the right ventricle (RVAD)) maintain or improve mechanical function of severely failing hearts by pumping the major portion of the blood volume. Transcriptome profiling indicates, however, that mechanical unloading by different VAD types (pulsatile and nonpulsatile VADs) does not evoke a gene activity pattern supportive of reverse remodeling [23]. Even though the VAD mechanically unloads the myocardium, the heart muscle might still work under conditions where the energy support does not match the energy demand. This may be one reason for the observed discrepancy between recovery of function and transcriptome profile. Any mechanical overload affects the gene expression profile. Under mechanical overload the gene expression profile displays a tendency to partly revert to the fetal expression program [10]. Although these changes are adaptive in order to cope with the increased workload and may be reversible upon mechanical support, beyond a certain limit they become irreversible precipitating myocardial failure. Probably most VADs are implanted at advanced disease stages when reversion to the normal is no longer possible.
Similar considerations also apply to the atrial artificial muscle device. It will be interesting to assess the gene expression pattern after application of the atrial nitinol support. The question whether the atrial support can reverse the gene expression programme towards normal or not, may depend on the time point of the disease course at which the device is applied. In early stages with atrial fibrillation (AF) as prominent symptome the altered gene expression may not have reached the point of no return. AF with high excitatory frequency imposes high energy consumption on the atrial wall. Lowering frequency and mechanical support at this early stage could restore gene expression together with the metabolic, Ca2+ handling, and electrical strains to a normal range close to a healthy heart.
Da capo
As opposed to conventional alloys, the nickel-titanium alloys (nitinol) are of commercial interest since they can recover from various deformations to the original crystal structure, a property called “shape memory” [18–20]. The “one-way shape memory effect” allows for one single movement against an externally set resistance as in self-expanding vascular stents, surgical suture clips, orthodontic brackets in dentistry, resilient glasses frames, temperature control systems, underwire bras, and many others. The design of “artificial muscle” adopts the principle of the “two-way shape memory effect” with nitinol oscillating between two types of crystal structures (as described above). Such devices are foreseen to support the function of insufficient natural muscles including the failing myocardium and to replace missing muscles altogether. To call these devices “artificial muscles” is, however, a euphemism if not a misnomer. Besides limited mechanical support the artificial nitinol muscles have nothing in common with natural muscles. In particular their contractile force cannot be smoothly regulated which is a fundamental property of natural movement with graded intensities. The mechanical impact is forced onto the natural muscles regardless of their actual condition with regard to metabolism, Ca2+-homeostasis and electrical sensitivity. It could be that the enforced mechanical impact on the gene expression profile further precipitates deterioration of the muscle to be treated. Taken together, the development of artificial muscle on the basis of nitinol wires is still in its infancy. Although a great number of applications can be imagined, the nitinol artificial muscle will have to prove its viability in the various clinical settings.
References
1 Tozzi P. Artificial muscle: the human chimera is the future. Swiss Med Wkly. 2011;141:w13311.
2 Huxley HE. Fifty years of muscle and sliding filament hypothesis. Eur J Biochem. 2004;271:1403–15.
3 Geeves MA, Holmes KC. Structural mechanism of muscle contraction. Annu Rev Biochem. 1999;68:687–728.
4 Finer JT, Simmons RM, Spudich JA. Single myosin molecule mechanics: piconewton forces and nanometre steps. Nature. 1994;368:113–9.
5 Linke WA. Sense and stretchability: the role of titin and titin-associated proteins in myocardial stress-sensing and mechanical dysfunction. Cardiovasc Res. 2008;77:637–48.
6 Dobson GP. On being the right size: heart design, mitochondrial efficiency and lifespan potential. Clin Exp Pharmacol Physiol. 2003;30:590–7.
7 Woledge RC, Curtin NA, Homsher E. Energetic aspects of muscle contraction. Academic Press, London 1985.
8 Gibbs CL, Loiselle DS. Cardiac basal metabolism. Jap J Physiol. 2001;51:399–426.
9 Schaub MC, Heizmann CW. Calcium, troponin, calmodulin, S100 proteins: from myocardial basics to new therapeutic strategies. Biochem Biophys Res Commun. 2008;369:247–64.
10 Schaub MC, Hefti MA, Zaugg M. Integration of calcium with the signaling network in cardiac myocytes. J Mol Cell Cardiol. 2006;41:183–214.
11 Ingwall JS, Weiss RG. Is the failing heart energy starved? On using chemical energy to support cardiac function. Circ Res. 2004;95:135–45.
12 Neubauer S. The failing heart – An engine out of fuel. N Engl J Med. 2007;356:1140–51.
13 Raichle ME, Gusnard DA. Appraising the brain’s energy budget. Proc Natl Acad Sci USA. 2002;99:10237–9.
14 Balaban RS. The role of Ca2+ signaling in the coordination of mitochondrial ATP production with cardiac work. Biochim Biophys Acta. 2009;1787:1334–41.
15 Beer M, Seyfarth T, Sandstede J, Landschütz W, Lipke C, Köstler H, et al. Absolute concentrations of high-energy phosphate metabolites in normal, hypertrophied, and failing human myocardium measured noninvasively with 31P-SLOOP magnetic resonance spectroscopy. J Am Coll Cardiol. 2002;40:1267–74.
16 Ingwall JS. Energy metabolism in heart failure and remodeling. Cardiovasc Res. 2009;81:412–9.
17 Ebron VH, Yang Z, Seyer DJ, Kozlov ME, Oh J, Xie H, et al. Fuel-powered artificial muscles. Science. 2006;311:1580–3.
18 Stoeckel D, Pelton A, Duerig T. Self-expanding nitinol stents: material and design considerations. Eur Radiol. 2004;14:292–301.
19 Dotter CT, Buschmann PAC, McKinney MK, Rösch J. Transluminal expandable nitinol coil stent grafting: preliminary report. Radiology. 1983;147:259–60.
20 Pelton AR, Russell SM, DiCello J. The physical metallurgy of nitinol for medical applications. J Minerals Metals Materials (JOM-US). 2003;55:33–7.
21 Tozzi P, Hayoz D, Taub S, Muradbegovic M, Rizzo E, et al. Biometal muscle to restore atrial transport function in a permanent atrial fibrillation animal model: a potential tool in the treatment of end-stage heart failure. Eur J Cardio Thorac Surg. 2010;37:870–4.
22 Neuberger HR, Mewis C, Van Veldhuisen DJ, Schotten U, Van Gelder IC, Allessie MA, et al. Management of atrial fibrillation in patients with heart failure. Eur Heart J. 2007;28:2568–77.
23 Schwientek P, Ellinghaus P, Steppen S, D’Urso D, Seewald M, Kassner A, et al. Global gene expression analysis in nonfailing and failing myocardium pre- and postpulsatile and nonpulsatile ventricular assist device support. Physiol Genomics. 2010;42:397–405.