Post genomic decade – the epigenome and exposome challenges
DOI: https://doi.org/10.4414/smw.2011.13321
Summary
Sequencing the human genome was the big challenge of the last decade. Ten years later, the large amount of DNA sequences accumulated in our databases allows us to look at genome variations between humans. The level of complexity of these variations is much higher than previously expected. It goes from changes in the nucleotidic sequence, such as single nucleotide polymorphisms (SNPs) or copy number variations (CNVs), to modifications in DNA transcription or methylation. Indeed, epigenetics, with chromatin modifications and underlying crosstalk between DNA methylation, histone tails acetylation and non coding RNAs, as microRNAs, all participate to this non-encoded gene expression regulation. Understanding the extent of genomic diversity between humans and linking it to phenotypes and diseases, unravelling the environmental exposures that may be detrimental for our health is the next challenge of the geneticists. The decrypting of the epigenome and the exposome is now on its way.
A genomic decade
Decoding the human genome
Humans have in common 99% of their DNA. The remaining 1% was believed to be responsible for all the interindividual differences: physical characteristics, disease risks, behaviour, and, in theory, all what makes each of us unique. After the launching of the Human Genome Project in 1990, the completion of the first individual genome sequences was reported in 2001 [1, 2] and the 1000 Genomes Project in 2010 [3, 4]. The latter project, an encyclopaedia of human genome variations is aimed at identifying all the single nucleotide polymorphisms (SNPs) found at a frequency of 1% or more in humans as well as other types of DNA variants, as genomic structural variants or insertions/deletions. Individuals’ DNAs from major ethnic groups (European, East Asian, South Asian, Americans and West Africans) were sequenced. Today’s sequencing machines can read approximately 250 billion bases per week, as compared to approximately 5 million in 2000 [5]. The 1000 Genomes Project was performed using the most cutting-edge and high-throughput genomic technology, and DNA from individual’s immortalised lymphoblastoid cell lines. It is a pilot study gathering data from 9 sequencing centres, with 4.9 trillion bases sequenced and a database of 15 million SNPs, among which more than half reported for the first time. In addition, the 1000 Genomes Project has identified 1 million DNA deletions or insertions as well as 20,000 other structural variations [3, 4]. However, in order to obtain a clearer understanding of how these variations contribute to human specific phenotypes, the functional pathways in which the corresponding genes are involved have to be elucidated. Then, the relative contribution of the variants to the phenotype or disease needs to be estimated.
Individual variations
Comparing each individual genome to the so called Reference Human Genome Map (a database genome cleared from all known mutations), it was found, surprisingly, that each individual may carry loss-of-function (LOF) mutations in 250 to 300 genes and may be heterozygous for 50 to 100 variants involved in inherited disorders. Therefore, beside polymorphisms, a huge amount of genetic changes was found to be present in the human genome showing that genetic perfection simply does not exist. Now, from data to understanding, the next step will be to correlate these LOF mutations to potential phenotypes, using a more traditional approach of cell to knock-out animal models, twin discordant for specific phenotypes and family studies. This will be a difficult task since LOF mutations can be found in genes of yet unknown function, may be present in gene regions skipped by alternative splicing or may be somatic mutations that are only present in a particular tissue, and not necessarily linked to a phenotype observed in another tissue [6]. The traditional candidate-gene approach has permitted, to date, to identify approximately 2850 genes underlying Mendelian diseases and 1100 loci involved in common polygenic diseases [5]. The whole genome sequencing technology will certainly allow the discovery of new dominant lethal disorders and new Mendelian diseases.
Copy Number Variations (CNVs) and Conserved non-Coding Elements (CNEs)
A surprising finding of the 1000 Genome Project was the occurrence in the genome of gene duplications and of variations in their numbers, among individuals, called Copy Number Variations (CNVs). The amplitude of this phenomenon, i.e., the number of gene copies, may be in association with modifications in protein production and therefore with phenotypic changes. Large differences in gene copy numbers were observed among different ethnic groups [3], by analysing 159 human genomes, with a sequencing coverage between 1.5 to 43 times for each genome, in order to decrease the risk of technical errors. Another study, the Human Genome diversity Project is documenting the genetic variations between human species worldwide, by sequencing DNA from 52 population subgroups spanning all continents [7]. Both projects and published results emphasise the need of sequencing an increasing number of individuals when trying to define rare variants (an estimation of 95% of variants defined for sequencing 1000 individuals), and to cover, or sequence several times, each sample, for the accuracy of genotypes results.
Another finding of the 1000 Genome Project was the occurrence in the genome of hundreds of conserved non-coding elements (CNEs). These genomic regions, outside the coding stretches of DNA, display high sequence conservation across various species, demonstrating that conservation is not restricted to the protein-coding regions, that indeed cover only the 1.5% of our genome [5].The CNEs should be of high functional importance, perhaps in association with specific phenotypes or diseases and the discovery of their existence increases the level of complexity of the gene-protein relationship.
RNA sequences
RNA sequencing has also been developed to unravel potential differences between the genome and the transcriptome. These RNA sequencing studies, for instance, revealed the existence, in numerous genes, of different forms of alternative splicing, resulting from the use or not of different exons of a gene unit [8]. This transcriptional diversity was emphasised by the recent results of the analysis of 18 genomes and the corresponding RNA transcript sequences of unrelated Korean individuals [9]. Extensive variations in the genomic and in the transcriptional profiles were reported, with more than 4,400 regions never annotated before as transcriptionally active, and 1809 sites where the transcriptome sequence did not exactly match the genomic sequence. Here again, the need of increasing the sequence read depth is critical, as well as the analysis of a variety of tissues and cell types.
Recently, the role of the different RNA types has been emphasised and grouped in an unifying theory of competing endogenous RNA (ceRNA) [10]. In the latter, microRNAs, non-coding RNAs, transcribed pseudogenes may be active partners and exert crossregulation of their expression levels. A whole transcriptome network, with multiple RNA partners, which may permit understanding of the complexity of an organism, as compared to its DNA code. Indeed, the discordance between the DNA and RNA sequences, i.e. the genome-transcriptome variations were analysed in a sample of 27 individuals, for a single cell type, and revealed thousands of exonic sites where the RNA sequence did not match the DNA sequence [11]. Therefore, the relation DNA-RNA-protein is not linear and RNA sequence variation should be taken into account as a player in human complexity.
At the individual level, the most recent technical advances and the elaboration of variant databases already allow comparing the entire human exome (i.e. the protein-coding regions of the entire genome) to reference sequences. These achievements represent most probably a first step to personalised sequencing and will permit analysing single-gene mutations and SNPs linked to increased risks for specific diseases. Personalised sequencing, within the next decade, is therefore expected to bring a major change in clinical practice. However, this type of approach is still controversial, as the risks estimated from the known genetic variants may account for only a small proportion of a specific disease-risk. The anxiety generated by the estimates of increased risks may also be highly detrimental. Finally, the risk of discrimination based on one’s genome variations cannot be ruled out.
From genomics to epigenomics
Non-nucleotidic modifications
In parallel with the huge advances of the genomic projects, it progressively appears that interindividual and interspecies diversity could not be due only to differences at the level of genetic sequences. Non-nucleotidic modifications of DNA leading to changes in gene expression, defined as epigenetic modifications, are now understood as a unique way to respond to the environment. They may explain the specific impact of an exposure to deleterious factors, on gene expression. RNAs and DNA-binded proteins, such as nucleosomes and histones, may be modified. Histone modifications (with several types of chemical changes such as phosphorylation or acetylation in nucleosomal histones), microRNA and DNA methylation are part of the epigenetic battery and add several layers of information and links to environmental changes. The spatial conformation of chromatin and accessibility to transcription factors is therefore variable. More than 50 different chromatin states have been defined and all these may have distinct biological consequences [12]. A yet unknown number of genes and of tissues (250 or more tissues for human) may indeed undergo specific epigenetic marking in response to different environmental exposures. The best characterised of these epigenetic markings is DNA methylation, that has been shown to lead to changes in chromatin structure and subsequently in gene expression (fig. 1). Methylation of occurs within CpG dinucleotides, on the fifth carbone of cytosine, and it is estimated that nearly 30 million CpGs in the human genome may be a target of methylation [13]. In addition, more than 100 potential histone modifications have been described. The combinations and functional consequences are yet unknown.
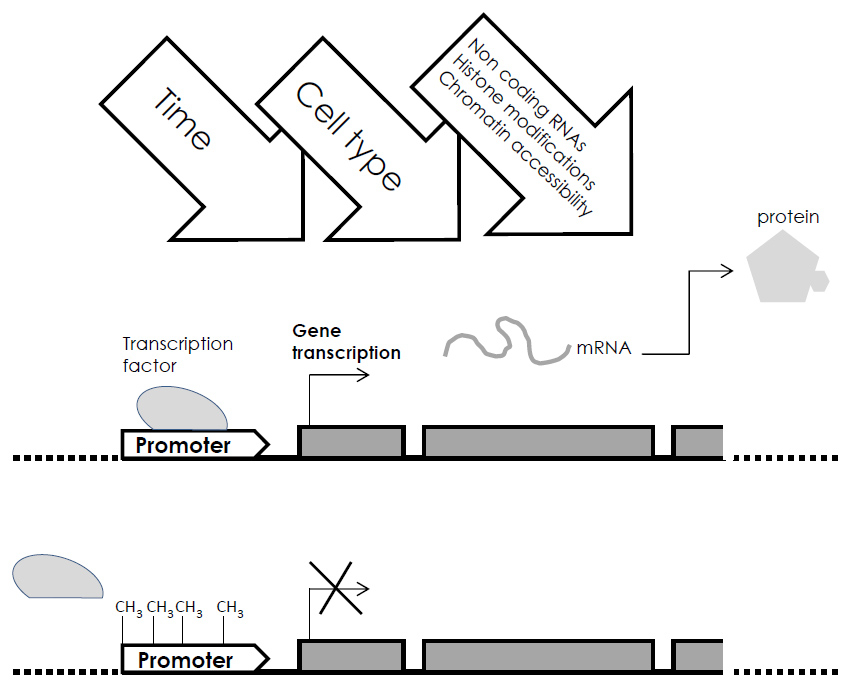
Figure 1
Representation of the methylation-gene silencing phenomenon. DNA methylation of specific sites in the gene’s promoter prevents the binding of the transcription factor and consequently gene transcription into mRNA and its subsequent expression. Cross-talking with time, cell-type and all the other epigenetic changes is represented.
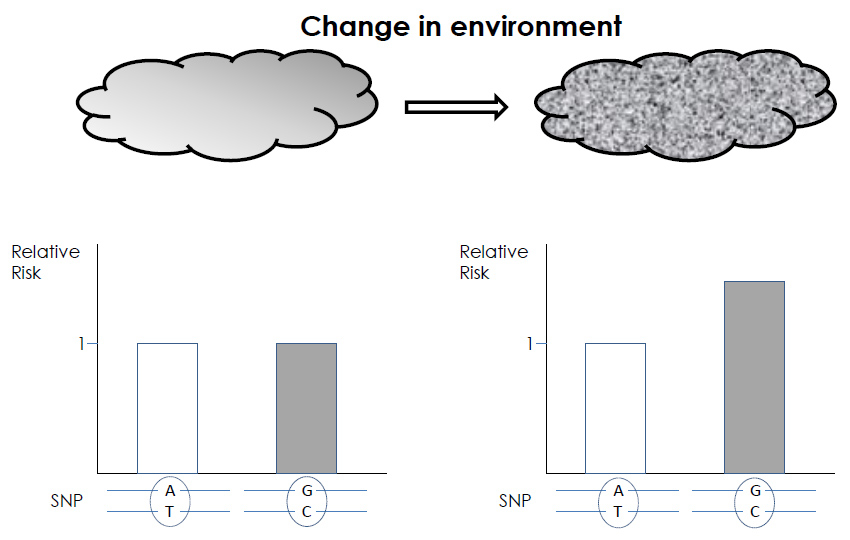
Figure 2
Two genotypes differing at a single SNP, with their relative contribution to the risk of developing a specific disease are shown (empty or gray columns). A change in the environment (from left to right graph) can reveal the role of a SNP in the risk of developing this disease: the theoretical risk of 1 is modified to 1.5.
Epigenome mapping
The development of new technologies for genome-wide DNA methylation analysis has started. It combines high-throughput sequencing methods with analysis of the global pattern of DNA methylation and chromatin modifications, at a single-base resolution level [14].
A large map of epigenomic variations now exists [15], that shows how the 1% interindividual DNA sequence differences can be amplified to result in large phenotypic or epiphenotypic variations. The International Human Epigenome Consortium (IHEC; http://www.ihec-epigenomes.org ) has set reference centres, with the goal of mapping hundreds of reference epigenomes within the next ten years. Methods, such as enrichment of methylated genomic DNA fragments or sequencing/pyrosequencing of bisulfite-converted DNA are being used for the genome-wide detection of methylated cytosines. Methylomic maps have already been achieved for some cell types, such as fetal and neonatal fibroblasts [15, 16]. Other methods and approaches have been developed, to target specific genomic regions, such as gene promoter regions [17]. However, an actual limitation to the interpretation of these methylomic maps is that they result from the analysis of a mixed cell population, each cell being indeed expected to have its own methylation status. A next step will also be to integrate this knowledge with the other regulation and epigenetic mechanisms. In parallel, epigenome-based therapies, such as de-methylating agents, are postulated, for the future prevention and treatment of epigenetic-related diseases. Again, the comparison between the methylation marks in healthy versus diseased cell types is a prerequisite to such developments.
Aggressions on the epigenome
When the epigenome is decrypted, the identification of all possible environmental deleterious factors, of their effects as a function of the period (pre- versus postnatal) and of the duration of the exposure, will be a very complex task. Furthermore, the epigenomic landscape changes with age, so that an individual epigenome does not remain the same across life. The prenatal period seems to to play a major role in the epigenome marking and may have a dramatically important incidence on adult health. The putatively predominant role of the in utero exposures is indeed referred to as the “developmental origin of adult disease” [18]. According to this view, epigenetic marking during fetal life would be a strong predictor of future diseases. Interestingly, a study reported that monozygotic twins, who are epigenetically nearly indistinguishable during the first years of life, develop later important differences in their epigenomic landscape [19]. This confirms that each individual develops his own somatic epigenetic marks during his life. The study of the effects of only a few key environmental factors, such as nutrition, behaviour, stress and toxicants, on only one type of epigenetic modification, such as methylation, is expected to yield, for any single individual, an enormous amount of data [20].
Putting together epigenetics and epidemiology data, at the level of population subgroups exposed to specific toxic compounds, or stressors, may allow bypassing a systematic and tricky approach like the epigenome mapping and should allow the links between an exposure to deleterious factors, a specific epigenetic marking, such as DNA methylation, and a disease to be demonstrated. However, the ability to understand, how DNA sequence variations together with the different epigenetic marking variations, play a role in the development of complex diseases will be a big challenge. Indeed, a SNP may, depending on the type of environmental exposure, give a different relative risk for a specific disease (fig. 2). Another level of complexity, for the interpretation of the epigenomic data, comes from the fact that a disease may, per se, induce modifications of the epigenetic marks, therefore masking the original pathogenic changes.
The exposome
The understanding of the complex interplay between the genome, the epigenome and the environment certainly needs to define another dimension: the exposome. This concept refers to the total exposures received during the organism’s life [21] and is primarily aimed at understanding key exposures linked to chronic diseases. It is currently estimated that the proportion of all human diseases that can be attributed to environmental pollutants, in addition to work exposure, is approximately 7 to 10% [22, 23]. On the other hand, the risks of cancer directly attributable to genetic factors is only 10%, as shown by studies on twins [24]. These numbers demonstrate that there is a real need to consider the environmental exposure as seriously involved in the pathophysiology of human diseases. Ideally, the exposome analysis should include the exposure not only to deleterious factors, such as toxic compounds, radiation, drugs, air and water pollution, but also to lifestyle factors such as diet, smoking and stress, from the prenatal period to death. The exposome is indeed a very variable and unstable entity that evolves throughout the lifetime and has to face the understanding of the impact of complex mixtures and of individual vulnerability, the understanding of the non – relevant exposures, as well as the definition of measurable biomarkers of exposure. Developing reliable measurement tools as well as a way of recording complete exposure histories is an extremely challenging task. It will necessitate important cohorts of individuals, with a longitudinal follow-up, detailed questionnaires and very large biobanks for the storage and analysis of the individual’s biological samples [25]. Critical life stages, such as fetal development, and early childhood, should be considered with particular attention [21]. Indeed, the National Children’s Study should provide data and biological samples on the first 21 years of life of a human prospective cohort [26]. It is now relatively easy to perform genome wide association studies (GWAS), by comparing a large number of genomes of affected and non affected individuals. The next step will be to perform environment wide association studies (EWAS) by comparing individual exposomes. A recent study addressed the respiratory exposome by measuring volatile chemicals produced normally by the metabolism and environmental compounds in exhaled breath samples of 130 volunteers [27]. Another approach, illustrated by the French national occupational disease surveillance and prevention network (RNV3P) consisted in collecting and analysing 58777 occupational health standardised reports to track possibly new exposure-disease associations [28]. The important amount of data collected in these studies clearly shows that, working on the exposome implies a collaborative effort, large consortiums of genomics, epigenomics and cutting-edge technological platforms. An important laboratory in vitro tool is the use of cell-culture model systems, permitting replication or diversified exposures and looking downstream at the gene expression profiles. From that, key environmental exposure involved in the development of human chronic diseases may be discovered and appropriate prevention and regulation developed. Figure 3 shows the interactions between these different approaches and the different parameters and variables analysed.

Figure 3
Epigenome, exposome and genome interactions with regards the development of multifactorial diseases. SNPs: Single Nucleotide Polymorphisms. CNVs: Copy Number Variations. PTMs: Posttranslational histone modifications (methylation, acetylation, phosphorylation and ubiquitilation).
Conclusion
What makes us, as an individual, at risk of developing a common disease? Nature and nurture. Top-down approaches (from biomarkers to the exposome) and bottom-up approach (from the exposome to biomarkers), With the genomic, epigenomic and exposomic projects we are moving fast forward, but an important task will be to develop a new ethic adapted to the new era of personalised medicine. An ethic-omics worldwide consortium?
References
1 Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, et al. The sequence of the human genome. Science. 2001;291(5507):1304–51.
2 Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921.
3 Sudmant PH, Kitzman JO, Antonacci F, Alkan C, Malig M, Tsalenko A, et al. Diversity of human copy number variation and multicopy genes. Science. 2010;330(6004):641–6.
4 Altshuler D, Durbin RM, Abecasis GR, Bentley DR, Chakravarti A, Clark AG, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061–73.
5 Lander ES. Initial impact of the sequencing of the human genome. Nature. 2011;470(7333):187–97.
6 MacArthur DG, Tyler-Smith C. Loss-of-function variants in the genomes of healthy humans. Hum Mol Genet. 2010;19(R2):R125–R130.
7 Cavalli-Sforza LL. The Human Genome Diversity Project: past, present and future. Nat Rev Genet. 2005;6(4):333–40.
8 Forrest AR, Carninci P. Whole genome transcriptome analysis. RNA Biol. 2009;6(2):107–12.
9 Ju YS, Kim JI, Kim S, Hong D, Park, Shin JY, et al. Extensive genomic and transcriptional diversity identified through massively parallel DNA and RNA sequencing of eighteen Korean individuals. Nat Genet. 2011;43(8):745–52.
10 Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146(3):353–8.
11 Li M, Wang IX, Li Y, Bruzel A, Richards AL, Toung JM, et al. Widespread RNA and DNA sequence differences in the human transcriptome. Science. 2011;333(6038):53–8.
12 Ernst J, Kellis M. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat Biotechnol. 2010;8:817–25.
13 Milosavljevic A. Emerging patterns of epigenomic variation. Trends Genet. 2011;6:242–50.
14 Ndlovu MN, Denis H, Fuks F. Exposing the DNA methylome iceberg. Trends Biochem Sci. 2011;36(7):381–7.
15 Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462(7271):315–22.
16 Laurent L, Wong E, Li G, Huynh T, Tsirigos A, Ong CT, et al. Dynamic changes in the human methylome during differentiation. Genome Res. 2010;20(3):320–31.
17 Weber M, Hellmann I, Stadler MB, Ramos L, Pääbo S, Rebhan M, et al. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet. 2007;39(4):457–66.
18 Barker DJ, Eriksson JG, Forsén T, Osmond C. Fetal origins of adult disease: strength of effects and biological basis. Int J Epidemiol. 2002;31(6):1235–9.
19 Fraga MF, Ballestar E, Paz MF, Ropero S, Setien, F, Ballestar ML, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A. 2005;102(30):10604–9.
20 Faulk C, Dolinoy DC. Timing is everything: the when and how of environmentally induced changes in the epigenome of animals. Epigenetics. 2011;6(7):791–7.
21 Rappaport SM. Implications of the exposome for exposure science. J Expo Sci Environ Epidemiol. 2011;21(1):5–9.
22 Rodgers A, Ezzati M, Vander Hoorn S, Lopez, AD, Lin RB, Murray CJ, et al. Distribution of major health risks: findings from the Global Burden of Disease study. PLoS Med. 2004;1(1):e27.
23 Saracci R, Vineis P. Disease proportions attributable to environment. Environ Health. 2007;6:38.
24 Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, et al. Environmental and heritable factors in the causation of cancer – analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000;343(2):78–85.
25 Wild CP. Complementing the genome with an “exposome”: the outstanding challenge of environmental exposure measurement in molecular epidemiology. Cancer Epidemiol Biomarkers Prev. 2005;14(8):1847–50.
26 Lioy PJ and Rappaport SM. Exposure science and the exposome: an opportunity for coherence in the environmental health sciences. Environ Health Perspect. 2011;119(11):a466-.
27 Pleil JD, Stiegel MA, Sobus JR. Breath biomarkers in environmental health science: exploring patterns in the human exposome. J Breath Res. 2011;5(4):046005.
28 Bonneterre V, Faisandier L, Bicout D, Bernardet C, Piollat J, Ameille J, et al. Programmed health surveillance and detection of emerging diseases in occupational health: contribution of the French national occupational disease surveillance and prevention network (RNV3P). Occup Environ Med. 2010;67(3):178–86.