The reception and the party after: how vascular endothelial growth factor receptor 2 explores cytoplasmic space
DOI: https://doi.org/10.4414/smw.2011.13318
P
Berger, K
Ballmer-Hofer
Summary
Vascular endothelial growth factors (VEGFs) regulate blood and lymph vessel formation through activation of the type V receptor tyrosine kinases VEGFR-1, -2 and -3. In addition, VEGFs interact with co-receptors such as neuropilins, integrins, semaphorins or heparansulfate glycosaminoglycans. Ligand binding dimerises the receptors and activates their intracellular tyrosine kinase domains, resulting in phosphorylation of tyrosine residues acting as docking sites for intracellular signalling molecules. Ligand-induced receptor is internalised and then transported through early, late, and recycling endosomes, and finally degraded by proteasomal or lysosomal pathways. Biological output by VEGF is mediated through distinct receptor/co-receptor complexes and generates signals in all cellular compartments triggering cellular responses such as cell migration, cell proliferation, vessel formation and maturation, as well as changes in vessel fenestration, constriction and permeability. Here we review recent experiments showing how VEGFR-2 is transported through intracellular vesicular compartments specified by Rab family GTPases, and discuss how different VEGF-A isoforms specify intracellular receptor trafficking. We also discuss how the biological consequences of aberrant receptor trafficking bear on the development of vascular disease.
Introduction to VEGF receptors
Signalling by receptor tyrosine kinases (RTKs) requires dimerisation with precise positioning of receptor subunits initiated and controlled by ligand binding. Dimeric ligand/receptor complexes subsequently initiate transmembrane signalling which results in activation of the intracellular tyrosine kinase domain [1, 2]. Vascular endothelial growth factors (collectively abbreviated here as ‘VEGF’) constitute a large family of angiogenic and lymphangiogenic polypeptides, VEGF-A, -B, -C, -D, -E, and -F, and placenta growth factor (PlGF) [3]. These ligands bind to specific RTKs, called VEGF receptors (VEGFR), and interact in an isoform-specific manner with additional cell surface-exposed proteins acting as co-receptors in angiogenic signalling. The biological functions of VEGF polypeptides predominantly arise from binding to the type V RTKs VEGFR-1 (Flt-1), VEGFR-2 (KDR/Flk-1), and VEGFR-3 (Flt-4) [4–6]. Neuropilin-1 and -2 (NRP-1, -2) [7], integrins [8], ephrin-B2 [9] and heparan sulfate proteoglycans (HSPG) [10] are at present the best characterised co-receptors. Some VEGFs interact with multiple receptors while others show very specific receptor binding properties. So, for instance, VEGF-A binds VEGFR-1 and -2, PlGF and VEGF-B are specific for VEGFR-1 [11, 12], while VEGF-E and most VEGF-F variants exclusively bind VEGFR-2 [13–15], and VEGF-C and -D bind VEGFR-2 and -3 as well as NRP-2 [16-18] (summarised in fig. 1). The complexity of VEGF signalling is further increased by the fact that some of the family members are processed posttranscriptionally and posttranslationally, giving rise to a bewildering number of isoforms with distinct receptor and extracellular matrix binding properties (reviewed in [3, 19]). VEGF-A, for instance, exists in more than 20 isoforms and we have shown recently that different splice variants of exon 8 alter co-receptor recruitment and thus processing of VEGFR-2 [20].
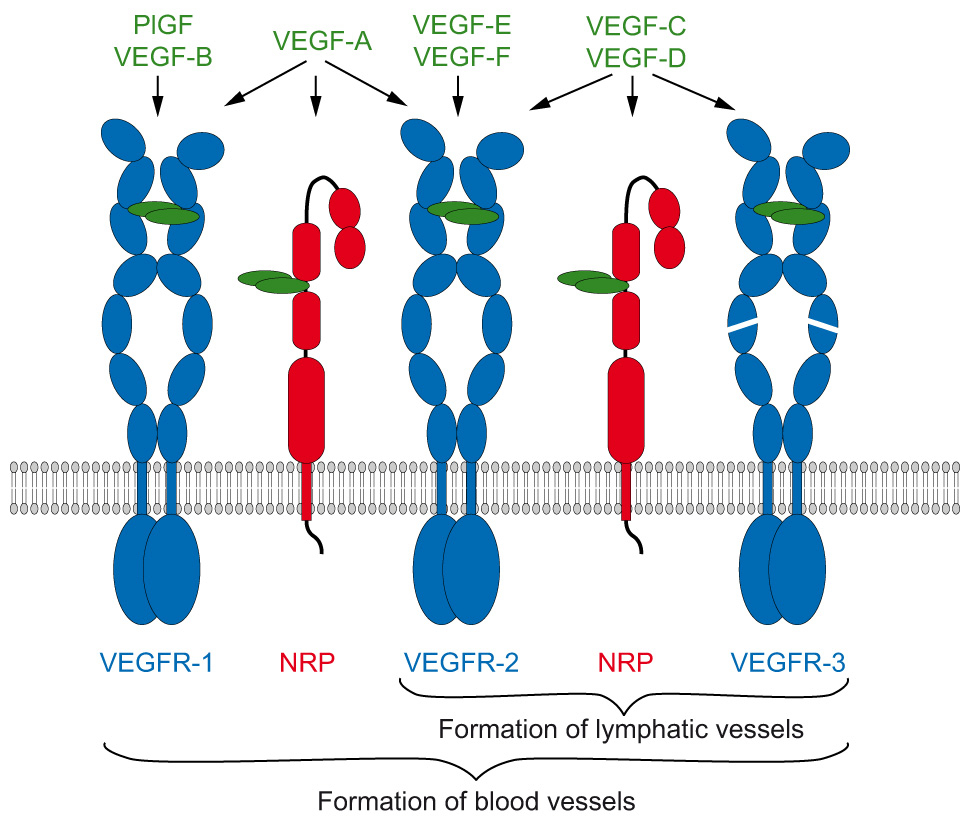
Figure 1
VEGFs and their interactions with VEGFRs and NRPs.
The human genome encodes five VEGF genes (A, B, C, D, and PlGF). Two VEGFs are encoded by viruses (E) and snakes (F), respectively. They all exist as dimers and induce dimerisation of the VEGFRs upon binding. Their binding specificity to the three VEGFRs and NRPs is indicated by arrows.
Ligand binding instigates transmembrane signalling and thereby generates distinct cellular output (reviewed in [21]). VEGFR-2 is the major receptor through which VEGFs regulate angiogenesis and vasculogenesis, and this receptor is essential for both blood and lymph vessel development and homeostasis. Distinct receptor/co-receptor complexes are formed upon activation with specific VEGF-A splice variants. The core domain of all VEGF-A variants encoded by exons 2-5 interacts with VEGFR-1 or -2, while sequences encoded by exons 6 and/or 7 and 8 determine co-receptor binding [22, 23]. Some VEGF variants bind simultaneously to two receptors such as VEGFR-2 and NRP-1 even when these are expressed separately on adjacent cells [24]. This might be required in order to promote endothelial cell migration and cell guidance, e.g. when vessels form along tracks defined by neural cells [25, 26] or during endothelial tip cell guidance [27, 28].
VEGFR-2 is internalised and transported to specific intracellular vesicular compartments upon ligand binding
Activation of VEGFRs initiates cell signalling at multiple steps of receptor processing in many distinct cellular compartments to promote endothelial cell survival, migration and proliferation as well as vessel fenestration and permeabilisation. Ligand-bound, activated receptors are transferred from the cell surface to intracellular vesicles and subsequently shuttled through early, late, and recycling endosomes. Activated receptors are destined for degradation in lysosomes or proteasomes, but evidence is emerging that subpopulations are recycled through the Golgi or directly back to the plasma membrane. Activated receptors initiate signalling in all cellular compartments during intracellular trafficking [29]. This documents that the cellular signalling machinery is compartmentalised to ensure proper spatio-temporal signal processing and integration.
Contrary to other RTKs, unstimulated VEGFR-2 is only partially localised at the plasma membrane with a significant fraction of the receptor present in internal vesicles [30, 31]. Inactive, unphosphorylated VEGFR-2 is present in resting cells in perinuclear caveolae which carry the early endosomal marker EEA-1 and dynamin [32]. Ligand-activated VEGFR-2 is rapidly internalised into early endosomes via clathrin coated vesicles. An indispensable step in receptor activation seems to be the release of receptors from adherens junctions [33] and caveolae [34]. More specifically, it was shown that VEGFR-2 is released from a lipid raft/caveolae membrane fraction and accumulates in focal adhesion contacts, where it becomes associated with a small subpopulation of caveolin phosphorylated at specific tyrosine residues following receptor activation [35]. This process is mediated by small G proteins of the Arf family.
Resting endothelial cells (EC) are organised in tightly associated cell monolayers held together by adherens junctions. Activation and release of VEGFR-2 from these structures and subsequent transfer to late endosomes, multi-vesicular bodies and finally lysosomes or proteasomes is achieved within minutes after ligand binding and leads to reorganisation of the cellular cytoskeleton and thus of EC monolayer morphology. This highly regulated process requires VEGFR-2 to interact with and activate a set of downstream signalling molecules which constitute the multiple molecular interactions regulating receptor trafficking to specific cellular compartments where biological signal output is generated. Phosphorylation of tyrosine residues 1054 and 1059 in the kinase activation loop stabilises receptor activity and is required for receptor internalisation [36]. Phosphorylation of tyrosine 951, 1175 and 1214 recruits and subsequently activates specific downstream signalling molecules mediating biological output [37]. Finally, VEGFR-2 is inactivated by dephosphorylation and recycled back to the plasma membrane or is degraded. Both proteasomal and lysosomal degradation have been described [38, 39]. A recent study showed that serine phosphorylation of a PEST motif in VEGFR-2 is essential for ubiquitination by the β-Trcp1 ubiquitin ligase and triggers proteasomal degradation [40]. The data describing the role in degradation of the classical E3 ligase c-Cbl, which regulates localisation and degradation of several RTKs, are conflicting [41]. VEGFR-2 is apparently directly ubiquinated, but it was also shown that ubiquitination of phospholipase C-γ1, one of the major targets of activated VEGFR-2, attenuates receptor signalling [41–43]. VEGF-A mediated activation of VEGFR-2 is also regulated by specific cleavage of the cytoplasmic domain [44]. The rate at which VEGFR-2 is degraded thus depends on several cellular parameters such as the status of adhesion junction complexes [45] and the association with co-receptors such as neuropilins [46], as discussed in more detail below.
Intracellular trafficking of VEGFR-2 and NRP-1, a closer look at the role of Rab GTPases
VEGFR-2 and NRP-1 remain membrane-bound on intracellular vesicles even after internalisation exposing their carboxyterminal domains to the cytoplasm. These receptors are then shuttled and sorted among the various intracellular membrane compartments of the cell. SNARE proteins are the major mediators of vesicle fusion and Rab GTPases (reviewed in [47]) are, together with phosphoinositides (reviewed in [48]), the coordinators of this membrane sorting process. Rab GTPases act as molecular switches recruiting distinct effector molecules in their active (GTP-bound) state [49–53]. These protein complexes then orchestrate the correct transfer of vesicles and their associated cargo among the different intracellular compartments in a unidirectional manner. Mutant forms of Rab GTPases have become popular tools for monitoring and manipulation of membrane trafficking of RTKs [54].
It was shown for EGFR that kinase activation results in conversion of Rab5 to its active form at the plasma membrane, a step that is essential for receptor internalisation [55]. Jopling et al. [56] were the first to show that VEGFR-2 co-localises with Rab5 and Rab7. They also showed that inactivation of Rab GTPases blocked receptor trafficking. Blocking Rab7 with a dominant negative mutant or with siRNA reduced phosphorylation of VEGFR-2 at tyrosine 1175 and resulted in increased p42/p44 MAPK signalling [56]. We recently showed that two different exon 8 splice variants of VEGF-A165 promote specific VEGFR-2 trafficking [20]. The NRP1-binding isoform VEGF-A165a led to receptor recycling to the plasma membrane through Rab11, while an isoform unable to bind NRP-1, VEGF-A165b, failed to do so. Interestingly, VEGFR-2 present in Rab 11 vesicles was not phosphorylated, showing that a so far undefined phosphatase regulates recycling of VEGFR-2 to the plasma membrane. Candidate phosphatases are DEP-1, PTP1B, SHP1, SHP2, and HCPTPA (see below).
The function of NRP-1 and GIPC/synectin in VEGFR-2 trafficking
Recruitment of NRP-1 into the VEGFR-2 complex by VEGF-A165a has significant implications for receptor recycling and phosphorylation as described above, but it is also relevant for signalling in vivo. It was previously shown that the NRP-1-binding ligand VEGF-A165a activates the p38 MAP kinase and promotes sprouting of intersegmental blood vessels in developing zebrafish in vivo. VEGF-A121, which has reduced affinity for NRP-1 and is unable to induce co-receptor complex formation, was unable to mediate these effects [22]. This was in agreement with the phenotype of NRP-1 null mice displaying severely impaired blood vessel formation in the developing embryo [25]. Similar defects were observed in zebrafish lacking NRP-1 [57]. Apparently the three carboxyterminal amino acids of NRP-1 constitute the PDZ domain binding motif interacting with GIPC, an adaptor molecule also known as synectin [58]. Ablation of GIPC in mice or zebrafish led to similar vascular defects as NRP-1 knockdown, establishing that these two genes functionally interact in vivo[59, 60]. Myosin VI is a well characterised interaction partner of GIPC and its function in endocytosis is well documented [61, 62]. Experiments with myosin VI null mice and knockdown experiments in zebrafish confirmed that this cytoskeletal protein plays a role in vessel development [63]. This suggests that myosin VI, together with NRP-1 and GIPC, are organised in a complex essential for angiogenesis by mediating internalisation of VEGFR-2 (fig. 2). Our data clearly show that the PDZ binding motif of NRP-1 is necessary for the Rab4 to Rab11 transition, and it was also shown that GIPC is present on Rab5 vesicles [20, 61]. We thus propose that the attachment of an NRP-1/GIPC/myosin VI complex to VEGFR-2 by VEGF A165a leads to association with the actin cytoskeleton soon after internalisation. It is unclear whether intact vesicles are then transported along the actin cytoskeleton or are disassembled by tubulation upon interaction with actin and myosin on endosomes [64]. It is unlikely that myosin VI is also responsible for the transport of VEGFR-2 back to the plasma membrane, since it is a minus-end-directed actin motor. Myosin VI and GIPC are presumably released from Rab11 vesicles and the resides are subsequently linked to a plus-end-directed myosin such as myosin Vb. In fact, it was shown that myosin Vb binds to Rab11 family-interacting protein-2 (Rab11-FIP2), an effector of Rab11 (fig. 2C). This ternary complex consisting of myosin Vb, Rab11-FIP2 and Rab11 could then initiate membrane recycling to the plasma membrane. The role of myosin Vb in angiogenesis has, however, not been determined so far [65].
Signalling during intracellular receptor trafficking
Binding of VEGF to VEGFR-2 results in receptor phosphorylation at several tyrosine residues located in the cytoplasmic receptor domain. To ensure proper receptor regulation and to prevent aberrant signalling, tyrosine phosphorylation of RTKs must be tightly regulated. The signal output also depends on the cellular context. Cadherins not only mediate cell-cell contacts, they also influence signalling [66]. VEGFR-2 co-localises with either caveolin-1 [34] or VE-cadherin at the plasma membrane in specific cellular contact sites called adherens junctions [33]. Endothelial adherens junctions contain catenins and the density-enhanced phosphatase-1 (DEP-1/CD148) as well as VEGFR-2 and VE-cadherin. VE-cadherin is an important modulator of VEGF signalling in vivo and was shown to negatively regulate VEGFR-2 activity [33, 67]. Silencing of DEP-1 increases VEGFR-2 internalisation and signalling, indicating that downregulation of receptor activity in the presence of VE-cadherin in resting EC monolayers is mediated by DEP-1. However, VEGFR-2 and VE-cadherin do not co-internalise and therefore DEP-1 may dephosphorylate VEGFR-2 exclusively at the plasma membrane.
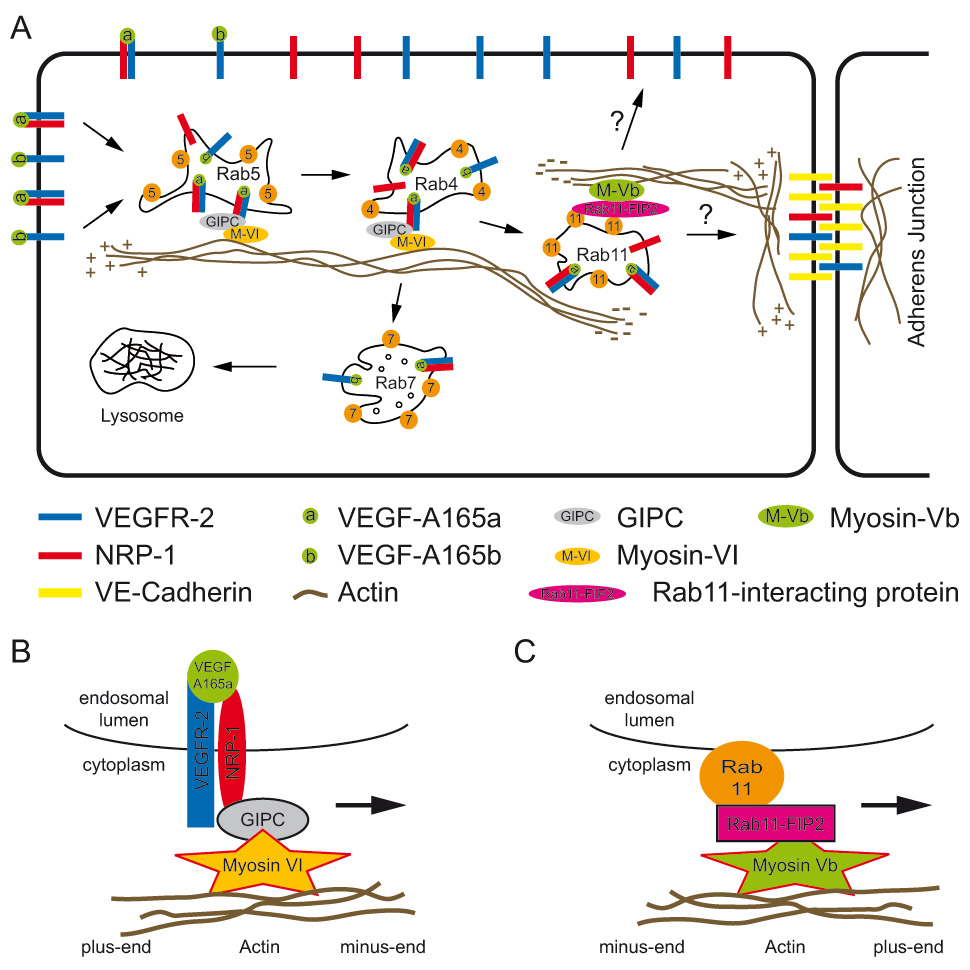
Figure 2
Influence of NRP-1 on VEGFR-2 trafficking.
(A) VEGF A165a links NRP-1 to VEGFR-2 leading to internalisation of the complex. NRP-1 rapidly recruits GIPC and myosin VI, a minus-end-directed actin motor. It is unclear how long this complex is active. We assume that it transports the complex to Rab11 vesicles since NRP-1 is necessary for the Rab4 to Rab11 transition. Recycling needs a plus-end-directed myosin motor such as for example myosin Vb, which indirectly interacts with Rab11. The insertion site at the plasma membrane is not known. The recycled factor can either contribute to the free VEGFR-2 pool on the plasma membrane or it can be inserted into adherens junctions. VEGFR-2 bound to VEGF A165b is preferentially routed via Rab5, Rab4, and Rab7 to lysosomes or proteasomes. (B) Detailed view of the minus-end-directed myosin complex. (C) Detailed view of a plus-end-directed myosin complex. Note that suggested role in angiogenesis of the complex shown is not proven yet.
T-cell protein tyrosine phosphatase, TCPTP, also known as PTPN2, is a non-receptor tyrosine phosphatase broadly expressed in most tissues. TCPTP is a negative regulator of many cancer-relevant signalling pathways and dephosphorylates RTKs such as PDGFR-β, EGFR, or VEGFR-2. It is activated by the cytoplasmic domain of integrin α1, indicating that TCPTP also acts at the plasma membrane, similarly to DEP-1. In focal adhesions, TCPTP dephosphorylates VEGFR-2 but does not dephosphorylate tyrosine 1175, suggesting that it differentially influences downstream signalling [68]. In endothelial cell monolayers held together by adherens junctions or integrin α1β1 basement membrane, VEGFR-2 is inactivated by DEP-1 and TCPTP. During angiogenesis, the basement membrane and cell-cell junctions are disrupted and activation of VEGFR-2 is no longer blocked by these phosphatases.
Overexpression of another protein tyrosine phosphatase, PTP1B dephosphorylating VEGFR-2 and VE-cadherin, results in reduced receptor phosphorylation and attenuation of ERK1/2 activity. As expected, knockdown of PTP1B by siRNA has the opposite effect. Interestingly, p38 activation is not affected under these conditions [69]. We assume that PTP1B acts early after internalisation and interfering with GIPC-myosin VI trafficking promotes prolonged exposure to PTP1B and thus reduces phosphorylation at tyrosine 1175, as shown by blockage of arteriogenesis. This is confirmed by the fact that PTP1B siRNA restores arterial morphogenesis [63].
Src-homology-2 domain-containing protein tyrosine phosphatase 2 (SHP-2, also known as PTPN11) is another non-receptor type tyrosine phosphatase involved in downregulation of VEGFR-2 signalling [70]. Sinah et al. [71] showed that activation of the dopamine receptor leads to activation of SHP-2, promoting dephosphorylation of VEGFR-2 at tyrosine Y951, Y996, and Y1059, but not at tyrosine Y1175, required for endothelial migration. The close homologue SHP-1 (also known as PTPN6) also negatively regulates VEGFR-2 phosphorylation, but with slightly different specificity [72]. Finally, HCPTPA is another low molecular weight tyrosine phosphatase involved in downregulating VEGFR-2 signalling [73] while VE-PTP dephosphorylates Tie-2 [74, 75] .
Future directions and medical applications
Besides Rab GTPases a particular type of lipids, phosphoinositides, which exist in cells in multiply-phosphorylated species localised in distinct subcellular membrane compartments, regulate vesicular protein transport. In addition, the late stage of protein trafficking is regulated by ESCRT proteins, which are involved in transferring proteins from late endosomes into multivesicular bodies and lysosomes [76]. Spatio-temporal regulation of cell signalling is the consequence of receptors being targeted to specific cellular compartments that carry specific signalling molecules. From what was presented here it is obvious that interfering with intracellular protein trafficking may represent a promising means of modifying signal output.
Pathological angiogenesis is a hallmark of many diseases and plays a role in tumour vascularisation, atherosclerosis [77], wound healing [78], cardiovascular disease [79], in diabetes [80] or in ocular retinopathies [81–83]. The idea of blocking the formation of new blood and lymphatic vessels to prevent tumour growth and metastasis was proposed many decades ago by the late Judah Folkman [84, 85]. Understanding the process of angiogenesis and vasculogenesis in molecular terms sets the stage for the development of new drugs aiming at modulating these processes, either positively in ischaemic tissues, e.g. in diabetic patients, or negatively to block excess vessel growth, such as in attempts to halt or reverse macular degeneration. Treatment of retinopathy patients with inhibitors of VEGF signalling emerged as a highly successful strategy while the original promises to block tumour vascularisation to prevent tumour progression and metastasis have not yet been fulfilled. The limited success of VEGF inhibition in tumour therapy was in the meantime attributed to upregulation of c-Met under hypoxic conditions favouring invasive growth [86]. Clinical trials with combined inhibition of VEGFR-2 and c-Met signalling are currently ongoing. In the future it will be important to improve anti-angiogenic drugs by making them more specific, and to allow for a more targeted approach, in particular to ensure that normal vessels are not affected and thereby avoid serious side effects. Along this line a promising approach may also be to deplete only specific VEGF isoforms such as the pro-angiogenic VEGF A165a, without affecting VEGF A165b levels that may be required to maintain the existing vasculature. This review of VEGFR signalling and trafficking will hopefully arouse new enthusiasm for the development of drugs interfering with intracellular receptor trafficking.
References
1 Hubbard SR. Juxtamembrane autoinhibition in receptor tyrosine kinases. Nat Rev Mol Cell Biol. 2004;5(6):464–71.
2 Schlessinger J. Signal transduction. Autoinhibition control. Science. 2003;300(5620):750–2.
3 Stuttfeld E, Ballmer-Hofer K. Structure and function of VEGF receptors. IUBMB Life. 2009;61(9):915–22.
4 Shibuya M, Yamaguchi S, Yamane A, Ikeda T, Tojo A, Matsushime H, et al. Nucleotide sequence and expression of a novel human receptor-type tyrosine kinase gene (flt) closely related to the fms family. Oncogene. 1990;5(4):519–24.
5 Terman BI, Carrion ME, Kovacs E, Rasmussen BA, Eddy RL, Shows TB. Identification of a new endothelial cell growth factor receptor tyrosine kinase. Oncogene. 1991;6(9):1677–83.
6 Pajusola K, Aprelikova O, Korhonen J, Kaipainen A, Pertovaara L, Alitalo R, et al. FLT4 receptor tyrosine kinase contains seven immunoglobulin-like loops and is expressed in multiple human tissues and cell lines. Cancer Res. 1992;52(20):5738–43.
7 Neufeld G, Kessler O, Herzog Y. The interaction of Neuropilin-1 and Neuropilin-2 with tyrosine-kinase receptors for VEGF. Adv Exp Med Biol. 2002;51581–90.
8 Somanath PR, Ciocea A, Byzova TV. Integrin and growth factor receptor alliance in angiogenesis. Cell Biochem Biophys. 2009;53(2):53–64.
9 Sawamiphak S, Seidel S, Essmann CL, Wilkinson GA, Pitulescu ME, Acker T, et al. Ephrin-B2 regulates VEGFR2 function in developmental and tumour angiogenesis. Nature. 2010;465(7297):487–91.
10 Tessler S, Rockwell P, Hicklin D, Cohen T, Levi BZ, Witte L, et al. Heparin modulates the interaction of VEGF165 with soluble and cell associated flk-1 receptors. J Biol Chem. 1994;269(17):12456–61.
11 Errico M, Riccioni T, Lyer S, Pisano C, Acharya KR, Persico GM, De FS. Identification of placental growth factor determinants for binding and activation of Flt-1 receptor. J Biol Chem. 2004;279(42):43929–39.
12 Olofsson B, Korpelainen E, Pepper MS, Mandriota SJ, Aase K, Kumar, et al. Vascular endothelial growth factor B (VEGF-B) binds to VEGF receptor-1 and regulates plasminogen activator activity in endothelial cells. Proc Natl Acad Sci USA. 1998;95(20):11709–14.
13 Lyttle DJ, Fraser KM, Fleming SB, Mercer AA, Robinson AJ. Homologs of vascular endothelial growth factor are encoded by the poxvirus orf virus. J Virol. 1994;68(1):84–92.
14 Mercer AA, Wise LM, Scagliarini A, McInnes CJ, Buttner M, Rziha HJ, et al. Vascular endothelial growth factors encoded by Orf virus show surprising sequence variation but have a conserved, functionally relevant structure. J Gen Virol. 2002;83(Pt 11):2845–55.
15 Wise LM, Ueda N, Dryden NH, Fleming SB, Caesar C, Roufail S, et al. Viral vascular endothelial growth factors vary extensively in amino acid sequence, receptor-binding specificities, and the ability to induce vascular permeability yet are uniformly active mitogens. J Biol Chem. 2003;278(39):38004–14.
16 Karpanen T, Heckman CA, Keskitalo S, Jeltsch M, Ollila H, Neufeld G, et al. Functional interaction of VEGF-C and VEGF-D with neuropilin receptors. FASEB J. 2006;20(9):1462–72.
17 Jussila L, Alitalo K. Vascular growth factors and lymphangiogenesis. Physiol Rev. 2002;82(3):673–700.
18 Takahashi H, Shibuya M. The vascular endothelial growth factor (VEGF)/VEGF receptor system and its role under physiological and pathological conditions. Clin Sci (Lond). 2005;109(3):227–41.
19 Harper SJ, Bates DO. VEGF-A splicing: the key to anti-angiogenic therapeutics? Nat Rev Cancer. 2008;8(11):880–7.
20 Ballmer-Hofer K, Andersson AE, Ratcliffe LE, Berger P. Neuropilin-1 promotes VEGFR-2 trafficking through Rab11 vesicles thereby specifying signal output. Blood. 2011;118(3):816–26.
21 Grünewald FS, Prota AE, Giese A, Ballmer-Hofer K. Structure-function analysis of VEGF receptor activation and the role of coreceptors in angiogenic signaling. Biochim Biophys Acta. 2010;1804(3):567–80.
22 Kawamura H, Li X, Goishi K, van Meeteren LA, Jakobsson L, Cébe-Suarez S, et al. Neuropilin-1 in regulation of VEGF-induced activation of p38MAPK and endothelial cell organization. Blood. 2008;112(9):3638–49.
23 Cébe-Suarez S, Pieren M, Cariolato L, Arn S, Hoffmann U, Bogucki A, et al. A VEGF-A splice variant defective for heparan sulfate and neuropilin-1 binding shows attenuated signaling through VEGFR-2. Cell Mol Life Sci. 2006;63(17):2067–77.
24 Cébe-Suarez S, Grünewald FS, Jaussi R, Li X, Claesson-Welsh L, Spillmann D, et al. Orf virus VEGF-E NZ2 promotes paracellular NRP-1/VEGFR-2 coreceptor assembly via the peptide RPPR. FASEB J. 2008;22(8):3078–86.
25 Kawasaki T, Kitsukawa T, Bekku Y, Matsuda Y, Sanbo M, Yagi T, et al. A requirement for neuropilin-1 in embryonic vessel formation. Development. 1999;126(21):4895–902.
26 Neufeld G, Cohen T, Shraga N, Lange T, Kessler O, Herzog Y. The Neuropilins: Multifunctional Semaphorin and VEGF Receptors that Modulate Axon Guidance and Angiogenesis. Trends Cardiovasc Med. 2002;12(1):13–9.
27 Gerhardt H, Golding M, Fruttiger M, Ruhrberg C, Lundkvist A, Abramsson A, et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol. 2003;161(6):1163–77.
28 Gerhardt H, Ruhrberg C, Abramsson A, Fujisawa H, Shima D, Betsholtz C. Neuropilin-1 is required for endothelial tip cell guidance in the developing central nervous system. Dev Dyn. 2004;231(3):503–9.
29 Miaczynska M, Pelkmans L, Zerial M. Not just a sink: endosomes in control of signal transduction. Curr Opin Cell Biol. 2004;16(4):400–6.
30 Scott A, Mellor H. VEGF receptor trafficking in angiogenesis. Biochem Soc Trans. 2009;37(Pt 6):1184–8.
31 Gampel A, Moss L, Jones MC, Brunton V, Norman JC, Mellor H. VEGF regulates the mobilisation of VEGFR-2/KDR from an intracellular endothelial storage compartment. Blood. 2006;108(8):2624–31.
32 Bhattacharya R, Kang-Decker N, Hughes DA, Mukherjee P, Shah V, McNiven MA, et al. Regulatory role of dynamin-2 in VEGFR-2/KDR-mediated endothelial signaling. FASEB J. 2005;19(12):1692–4.
33 Lampugnani MG, Orsenigo F, Gagliani MC, Tacchetti C, DeJana E. Vascular endothelial cadherin controls VEGFR-2 internalization and signaling from intracellular compartments. J Cell Biol. 2006;174(4):593–604.
34 Labrecque L, Royal I, Surprenant DS, Patterson C, Gingras D, Beliveau R. Regulation of vascular endothelial growth factor receptor-2 activity by caveolin-1 and plasma membrane cholesterol. Mol Biol Cell. 2003;14(1):334–47.
35 Ikeda S, Ushio-Fukai M, Zuo L, Tojo T, Dikalov S, Patrushev NA, et al. Novel role of ARF6 in vascular endothelial growth factor-induced signaling and angiogenesis. Circ Res. 2005;96(4):467–75.
36 Dougher M, Terman BI. Autophosphorylation of KDR in the kinase domain is required for maximal VEGF-stimulated kinase activity and receptor internalization. Oncogene. 1999;18(8):1619–27.
37 Koch S, Tugues S, Li X, Gualandi L, Claesson-Welsh L. Signal transduction by vascular endothelial growth factor receptors. Biochem J. 2011;437(2):169–83.
38 Ewan LC, Jopling HM, Jia H, Mittar S, Bagherzadeh A, Howell GJ, et al. Intrinsic tyrosine kinase activity is required for vascular endothelial growth factor receptor 2 ubiquitination, sorting and degradation in endothelial cells. Traffic. 2006;7(9):1270–82.
39 Murdaca J, Treins C, Monthouel-Kartmann MN, Pontier-Bres R, Kumar S, Van Obberghen E, et al. Grb10 prevents Nedd4-mediated vascular endothelial growth factor receptor-2 degradation. J Biol Chem. 2004.
40 Meyer RD, Srinivasan S, Singh AJ, Mahoney JE, Gharahassanlou KR, Rahimi N. PEST motif serine and tyrosine phosphorylation controls vascular endothelial growth factor receptor 2 stability and downregulation. Mol Cell Biol. 2011;31(10):2010–25.
41 Singh AJ, Meyer RD, Navruzbekov G, Shelke R, Duan L, Band H, et al. A critical role for the E3-ligase activity of c-Cbl in VEGFR-2-mediated PLCgamma1 activation and angiogenesis. Proc Natl Acad Sci USA. 2007;104(13):5413–8.
42 Meyer RD, Husain D, Rahimi N. c-Cbl inhibits angiogenesis and tumor growth by suppressing activation of PLCgamma1. Oncogene. 2011;30(19):2198–206.
43 Rahimi N. A role for protein ubiquitination in VEGFR-2 signalling and angiogenesis. Biochem Soc Trans. 2009;37(Pt 6):1189–92.
44 Bruns AF, Herbert SP, Odell AF, Jopling HM, Hooper NM, Zachary IC, et al. Ligand-Stimulated VEGFR2 Signaling is Regulated by Co-Ordinated Trafficking and Proteolysis. Traffic. 2010;11(1):161–74.
45 Lampugnani MG, DeJana E. Adherens junctions in endothelial cells regulate vessel maintenance and angiogenesis. Thromb Res. 2007;120(Suppl 2):S1–S6.
46 Holmes DI, Zachary IC. Vascular endothelial growth factor regulates Stanniocalcin-1 expression via Neuropilin-1-dependent regulation of KDR and synergism with fibroblast growth Factor-2. Cell Signal. 2008;20(3):569–79.
47 Mosesson Y, Mills GB, Yarden Y. Derailed endocytosis: an emerging feature of cancer. Nat Rev Cancer. 2008;8(11):835–50.
48 Balla T, Szentpetery Z, Kim YJ. Phosphoinositide signaling: new tools and insights. Physiology (Bethesda ). 2009;24231–44.
49 Zerial M, McBride H. Rab proteins as membrane organizers. Nat Rev Mol Cell Biol. 2001;2(2):107–17.
50 De Renzis S, Sonnichsen B, Zerial M. Divalent Rab effectors regulate the sub-compartmental organization and sorting of early endosomes. Nat Cell Biol. 2002;4(2):124–33.
51 Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol. 2009;10(8):513–25.
52 Grosshans BL, Ortiz D, Novick P. Rabs and their effectors: achieving specificity in membrane traffic. Proc Natl Acad Sci USA. 2006;103(32):11821–7.
53 Jordens I, Marsman M, Kuijl C, Neefjes J. Rab proteins, connecting transport and vesicle fusion. Traffic. 2005;6(12):1070–7.
54 Zwang Y, Yarden Y. Systems Biology of Growth Factor-Induced Receptor Endocytosis. Traffic. 2008;10(4):349–63.
55 Barbieri MA, Roberts RL, Gumusboga A, Highfield H, Alvarez-Dominguez C, Wells A, et al. Epidermal growth factor and membrane trafficking. EGF receptor activation of endocytosis requires Rab5a. J Cell Biol. 2000;151(3):539–50.
56 Jopling HM, Odell AF, Hooper NM, Zachary IC, Walker JH, Ponnambalam S. Rab GTPase regulation of VEGFR2 trafficking and signaling in endothelial cells. Arterioscler Thromb Vasc Biol. 2009;29(7):1119–24.
57 Lee P, Goishi K, Davidson AJ, Mannix R, Zon L, Klagsbrun M. Neuropilin-1 is required for vascular development and is a mediator of VEGF-dependent angiogenesis in zebrafish. Proc Natl Acad Sci USA. 2002;99(16):10470–5.
58 Cai H, Reed RR. Cloning and characterization of neuropilin-1-interacting protein: a PSD-95/Dlg/ZO-1 domain-containing protein that interacts with the cytoplasmic domain of neuropilin-1. J Neurosci. 1999;19(15):6519–27.
59 Chittenden TW, Claes F, Lanahan AA, Autiero M, Palac RT, Tkachenko EV, et al. Selective regulation of arterial branching morphogenesis by synectin. Dev Cell. 2006;10(6):783–95.
60 Wang L, Mukhopadhyay D, Xu X. C terminus of RGS-GAIP-interacting protein conveys neuropilin-1-mediated signaling during angiogenesis. FASEB J. 2006;20(9):1513–5.
61 Aschenbrenner L, Lee T, Hasson T. Myo6 facilitates the translocation of endocytic vesicles from cell peripheries. Mol Biol Cell. 2003;14(7):2728–43.
62 Naccache SN, Hasson T, Horowitz A. Binding of internalized receptors to the PDZ domain of GIPC/synectin recruits myosin VI to endocytic vesicles. Proc Natl Acad Sci USA. 2006;103(34):12735–40.
63 Lanahan AA, Hermans K, Claes F, Kerley-Hamilton JS, Zhuang ZW, Giordano FJ, et al. VEGF Receptor 2 Endocytic Trafficking Regulates Arterial Morphogenesis. Dev Cell. 2010;18(5):713–24.
64 Puthenveedu MA, Lauffer B, Temkin P, Vistein R, Carlton P, Thorn K, et al. Sequence-dependent sorting of recycling proteins by actin-stabilized endosomal microdomains. Cell. 2010;143(5):761–73.
65 Hales CM, Vaerman JP, Goldenring JR. Rab11 family interacting protein 2 associates with Myosin Vb and regulates plasma membrane recycling. J Biol Chem. 2002;277(52):50415–21.
66 Resink TJ, Philippova M, Joshi MB, Kyriakakis E, Erne P. Cadherins and cardiovascular disease. Swiss Med Wkly. 2009;139(9-10):122–34.
67 Carmeliet P, Lampugnani MG, Moons L, Breviario F, Compernolle V, Bono F, et al. Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell. 1999;98(2):147–57.
68 Mattila E, Auvinen K, Salmi M, Ivaska J. The protein tyrosine phosphatase TCPTP controls VEGFR2 signalling. J Cell Sci. 2008;121(Pt 21):3570–80.
69 Nakamura Y, Patrushev N, Inomata H, Mehta D, Urao N, Kim HW, et al. Role of protein tyrosine phosphatase 1B in vascular endothelial growth factor signaling and cell-cell adhesions in endothelial cells. Circ Res. 2008;102(10):1182–91.
70 Mitola S, Brenchio B, Piccinini M, Tertoolen L, Zammataro L, Breier G, et al. Type I Collagen Limits VEGFR-2 Signaling by a SHP2 Protein-Tyrosine Phosphatase-Dependent Mechanism 1. Circ Res. 2005
71 Sinha S, Vohra PK, Bhattacharya R, Dutta S, Sinha S, Mukhopadhyay D. Dopamine regulates phosphorylation of VEGF receptor 2 by engaging Src-homology-2-domain-containing protein tyrosine phosphatase 2. J Cell Sci. 2009;122(Pt 18):3385–92.
72 Bhattacharya R, Kwon J, Wang E, Mukherjee P, Mukhopadhyay D. Src homology 2 (SH2) domain containing protein tyrosine phosphatase-1 (SHP-1) dephosphorylates VEGF Receptor-2 and attenuates endothelial DNA synthesis, but not migration*. J Mol Signal. 2008;38.
73 Huang L, Sankar S, Lin C, Kontos CD, Schroff AD, Cha EH, et al. HCPTPA, a protein tyrosine phosphatase that regulates vascular endothelial growth factor receptor-mediated signal transduction and biological activity. J Biol Chem. 1999;274(53):38183–8.
74 Mellberg S, Dimberg A, Bahram F, Hayashi M, Rennel E, Ameur A, et al. Transcriptional profiling reveals a critical role for tyrosine phosphatase VE-PTP in regulation of VEGFR2 activity and endothelial cell morphogenesis. FASEB J. 2009;23(5):1490–502.
75 Nottebaum AF, Cagna G, Winderlich M, Gamp AC, Linnepe R, Polaschegg C, et al. VE-PTP maintains the endothelial barrier via plakoglobin and becomes dissociated from VE-cadherin by leukocytes and by VEGF. J Exp Med. 2008;205(12):2929–45.
76 Hanson PI, Shim S, Merrill SA. Cell biology of the ESCRT machinery. Curr Opin Cell Biol. 2009;21(4):568–74.
77 Celletti FL, Waugh JM, Amabile PG, Brendolan A, Hilfiker PR, Dake MD. Vascular endothelial growth factor enhances atherosclerotic plaque progression. Nat Med. 2001;7425–9.
78 Bao P, Kodra A, Tomic-Canic M, Golinko MS, Ehrlich HP, Brem H. The role of vascular endothelial growth factor in wound healing. J Surg Res. 2008;15(2):347–58.
79 Flammer AJ, Luscher TF. Three decades of endothelium research: from the detection of nitric oxide to the everyday implementation of endothelial function measurements in cardiovascular diseases. Swiss Med Wkly. 2010;140w13122.
80 Rivard A, Silver M, Chen D, Kearney M, Magner M, Annex B, et al. Rescue of diabetes-related impairment of angiogenesis by intramuscular gene therapy with adeno-VEGF. Am J Pathol. 1999;154(2):355–63.
81 Ferrara N. Vascular endothelial growth factor and age-related macular degeneration: from basic science to therapy. Nat Med. 2010;16(10):1107–11.
82 Gariano RF, Gardner TW. Retinal angiogenesis in development and disease. Nature. 2005;438(7070):960–6.
83 Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473(7347):298–307.
84 Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;2851182–6.
85 Folkman J. Anti-angiogenesis: new concept for therapy of solid tumors. Ann Surg. 1972;175409–16.
86 You WK, Sennino B, Williamson CW, Falcon B, Hashizume H, Yao LC, et al. VEGF and c-Met blockade amplify angiogenesis inhibition in pancreatic islet cancer. Cancer Res. 2011;71(14):4758–68.