Figure 1
DOI: https://doi.org/10.4414/smw.2011.13290
Inflammatory bowel disease (IBD) is a group of inflammatory conditions of the small intestine and colon affecting 1.4 million people in the United States. The major types of IBD are Crohn’s disease (CD) and ulcerative colitis (UC). Although the precise nature of pathogenesis is not clearly understood, evidence indicates that a genetic predisposition of multiple genes and environmental factors are involved [1–6].
Currently over 50 IBD-associated genes/loci have been identified [7–15]. Fine mapping of a linkage peak on chromosome 10q23 initially identified multiple variants associated with CD and UC in the DLG5gene (Discs large Homolog 5; MIM 604090) [16]. DLG5, a member of the membrane associated guanylate kinase (MAGUK) family of proteins, known to form scaffolds for proteins involved in intracellular signal transduction, maintenance of cell junctions, and clustering of channel proteins at the cell surface [17, 18]. As this protein may be involved in maintenance of epithelial permeability, it is a plausible candidate for involvement in IBD.
The involvement of DLG5 variants in the pathogenesis of IBD remains unclear. The DLG5 variant R30Q (rs1248696), which changes amino acid 30 in exon 3 from arginine to glutamine, has been associated with IBD [16, 19–22]. However, several studies have failed to replicate the original associations of the R30Q allele with increased risk [23–31]. Another nonsynonymous SNP P1371Q (rs2289310 c. C4136A) has also been associated with IBD in two studies [16, 20], but this association has not been replicated in other studies [19, 26–28]. Differences in population allele frequencies could explain the lack of replication of the associations with IBD.
Using a familial and sporadic IBD population from central Pennsylvania, USA, we recently confirmed the genetic association of R30Q with IBD [32]. In the present study, we examined another 3 nonsynonymous DLG5 SNPs, replicated the association of P1371Q with IBD, and revealed a genetic interaction between R30Q and P1371Q in IBD susceptibility using a newly developed statistical model for epistasis analysis [33].
The patient cohort is the same as the one previously analysed [32].A total of 212 patients were studied, including:
– 106 patients (CD: 58 and UC: 47) from 58 families recruited for the Milton S Hershey familial IBD registry. In the registry, at least two family members were affected by IBD per family. The central Pennsylvania area is largely populated by immigrants from Western Europe (Germany, the Netherlands) and the United Kingdom, and thus presents itself as having a patient population with a relatively homogeneous genetic background. The age of these patients ranged from 16–92 (mean 51) years. Blood was collected from these study participants and used to derive B cell lines by Epstein Barr virus (EBV) transformation as previously described [35].
– 107 sporadic IBD patients (57 CD and 50 UC) were recruited from the Milton S Hershey Medical Center. The age of these patients ranged from 22–66 (mean 48) years. DNA from this group was obtained from blood or intestinal tissues harvested at the time of surgery.
– A total of 139 individuals without IBD from 58 families from the Milton S Hershey Familial IBD Registry were used as non-IBD controls. The age of these patients ranged from 16–92 (mean 51) years. Blood collected from the participants was used to derive B cell lines by EBV transformation as a source of DNA for genotype analyses.
– Unrelated healthy controls (n = 170) were obtained from the Milton S Hershey Medical Center (110 blood samples) and from the Philadelphia Gift of Life Donor Program (60 lung tissue samples), a geographically close population group. The unrelated healthy controls age range was 15–81 (mean 37) years.
All human tissues described above were approved by the Human Subjects Protection Offices of The Pennsylvania State University College of Medicine, and were undertaken with the understanding and written consent of each subject.
Genomic DNA from B cell lines was isolated with the QIAamp DNA blood kit (Qiagen Inc. Valencia, CA), and DNA from tissues (blood, intestine, and lung) was isolated with the QIAamp DNA Mini Kit (Qiagen Inc. Valencia, CA) according to the manufacturer’s instruction. DNA concentrations was determined with a NanoDrop ND-1000 spectrophotometer (NanoDrop Technology, Wilmington, DE), and stored at –70 °C until use.
Genotyping was performed using PCR–based restriction fragment length polymorphism (RFLP) and converted RFLP (cRFLP) methods. Detailed methods for genotyping DLG5 variants R30Q (G113A, rs1248696) and G1066G (C3222T, rs1248634) were described in our previous publications [32, 35]. For this study we additionally genotyped DLG5 nonsynonymous SNPs P1371Q, E514Q, and P979L. For P1371Q (C4136A, rs2289310) genotyping, a 117 bp DNA fragment was amplified from genomic DNA with PCR primers 23DLG5f (5’-GCACCACCACCCCGGAGtA-3’) and 14DLG5r (5-TGGAGCTGCTCTGCTTGGaT-3’). In the primer 23DLG5f, t is a mismatch to nucleotide C in order to destroy an internal FokI restriction site. In the primer 14DLG5r, a is a mismatch to nucleotide C, converting the SNP to a FokI restriction site. 100 ng of genomic DNA was used for PCR in a 30 µl volume. The PCR profile was as follows: 95 °C for 2 min, 5 cycles of 95 °C for 30 sec, 50 °C for 1 min, and 72 °C for 1 min, then 30 cycles of 95 °C for 30 sec, 58 °C for 1 min, and 72 °C for 1 min, followed by a final extension step at 72 °C for 4 min. PCR products (5 µl) were digested with FokI (New England Biolabs, Ipswich, MA) according to manufacturer’s instructions. Digested allelic products were separated on 8% PAGE and visualized using ethidium bromide staining. The C allele generated a PCR product of 117 bp, while the A allele generated two products, 97 and 20 bp. SNPs E514Q and P979L were genotyped using natural Taq and Nae RFLP respectively.
For genetic association study,Pearson’s X 2-test with one degree of freedom for allelic association was performed using Haploview (MIT/Harvard Broad Institute). We calculated genotype-based Odds Ratio (OR) using Fisher’s exact test on the contingency tables, and the effect of the association was reported with the corresponding p-values. A genotype difference was considered significant whenp <0.05.
We used a new general model for testing high-order epistatic interactions in complex disease [33]. This model not only allows the testing of additive and dominant effects at single SNPs, but is also able to detect four types of epistatic interactions, namely additive × additive, additive × dominant, dominant × additive, and dominant × dominant, between different SNPs in a case-control study. In brief, considering two SNPs A and B and the nine resulting genotypes (AABB, AABb, AAbb, AaBB, AaBb, Aabb, aaBB, aaBb, and aabb) in both cases and controls, the model can test epistatic interactions of different kinds from these nine genotype groups of observations. Simulation studies to investigate the statistical behaviour of the new model suggest that the model has good power and low false positive rates for a modest sample size (200 cases and 200 controls) [33]. In our present study, we tested each of these epistatic interactions for all possible pairs of five SNPs.
From the Ensembl genomic database ( http://www.ensembl.org ; Vega Transcript ID OTTHUMT00000048900), we found 11 genetic variations in the DLG5 cDNA sequences; 3 SNPs in 3’UTR and 8 in coding region. We previously genotyped two DLG5 SNPs, R30Q and G1006G, and found an association of R30Q but not G1006G with IBD [32]. In this study we genotyped and analysed three additional nonsynonymous DLG5 SNPs, E514Q, P979L, and P1371Q. Statistical analysis (table 1) showed that the A allele of P1371Q was significantly associated with IBD in case (IBD patients)-control (unrelated healthy individuals) analysis (OR = 2.335, 95% CI = 1.097–4.972, p = 0.0246). In the studied samples, the genotype of all individuals carrying the P1371Q A allele was CA; no homozygous AA individuals were found. The frequency of the A allele carriers in the sporadic IBD patients was 9.4% and in the unrelated healthy controls 5.9%, indicating an increased incidence of the mutation allele in IBD patients. However, the frequency of A allele carriers in the familial IBD registry was 10.6%, as high as the sporadic IBD patients, compared to the case control population (5.9%), therefore association of the A allele with IBD in the family members (members affected with IBD vs. members without IBD) was very weak (OR = 1.665, 95% CI = 0.771–3.597, p = 0.1908). E514Q and P979L were not found to be associated with IBD (this study; data not shown). As we reported in our previous paper [32], there was no statistically significant difference between diseased and non–diseased individuals for G1066G. Considering G1066G does not change encoded amino acid residue, it is likely that G1066G has no impact on IBD.
Figure 1
The gender effect on genetic association of R30Q with IBD has been observed in several studies, including our previous report [21, 30, 32, 34, 36]. The results from the present study, as shown in table 2, indicate that the A allele of P1371Q was associated with IBD only in females (OR = 3.765, 95% CI = 1.307–10.85, p = 0.0095), and not in males (OR = 1.141, 95% CI = 0.383–3.400, p = 0.8135). The patients studied here are the same as in our previous study, and this observation on P1371Q contrasts with that on R30Q, in which the A allele R30Q was associated with IBD only in men, but not in women [32]. According to the literature, the gender effect on genetic association of DLG5 with IBD is complicated. R30Q has been associated with IBD in men [22, 32, 34]. In other studies, R30Q was associated with a small reduction in risk of CD in women [30]. To further elucidate the gender effect of DLG5 on IBD, additional studies in other populations, as well as biological functional investigation are needed.
Merging the genotype data for P1371Q from this study with our previous genotype data for R30Q, we studied the epistatic SNP–SNP interaction between P1371Q and R30Q for association with IBD. The results are shown in table 3. Individuals with the A allele at P1371Q or the A allele at R30Q vs. all other individuals showed an increased risk of IBD (OR = 2.265, 95% CI = 1.405–3.652, p = 0.0007). This epistatic effect is more significant compared to the effects from an individual SNP (OR = 2.335, 95% CI = 1.097–4.972, p = 0.0246) for P1371Q, and OR = 2.131, 95% CI = 1.233–3.684, p = 0.0061) for R30Q (table 2).
As we described above, the association of P1371Q with IBD is not significant when comparing IBD patients to non–IBD family members in the familial IBD registry (table 1). We reported a similar result for R30Q [32]. However, analysis indicates that interaction between P1371Q and R30Q significantly affects the genetic association of DLG5 with IBD in the familial IBD registry (OR = 1.805, 95% CI = 1.126–2.897, p = 0.0138) (table 3). R30Q displayed a significant genetic effect on the disease according to the chi-square test (p = 0.0158) after adjusting for multiple tests by the conservative Bonferroni correction. Further analysis was made on two different components of genetic effects for this R30Q. R30Q showed significant interactions with P1371Q, involving a dominant x dominant effect (p = 0.006–0.050) after the Bonferroni correction. We found that the dominant effect of R30Q was significant (p = 0.010), whereas its additive effect was not significant.
Furthermore, we considered the A allele of R30Q as an IBD risk mutation for men (A30male) and the A allele of P1371Q as an IBD risk mutation for women (A1371female). The combination analysis (table 3) indicates that the association of A30male and A1371female is highly significant for sporadic IBD (OR = 4.311, 95% CI = 2.101–8.846, p <0.0001). Even in the familial IBD registry in which the allele A frequency is as high as the sporadic IBD group, a significant association with IBD was also observed (OR = 2.860, 95% CI = 1.380–5.925, p = 0.0034).
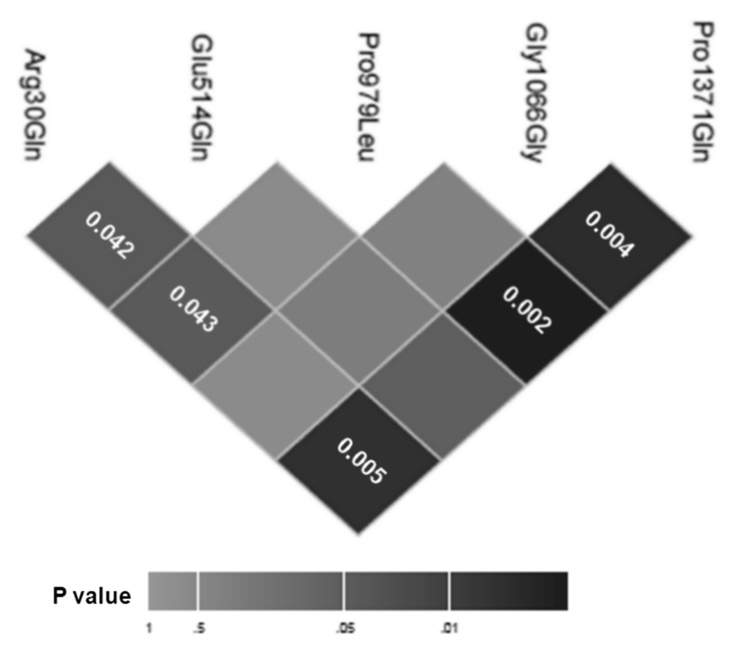
To study the interaction between 5 nonsynonymous DLG5 SNPs (R30Q and G1066G genotyped in previous paper [32]), we used a newly developed epistasis analysis method [33]. Figure 1 graphically depicts the epistatic interaction effects between homozygous genotype for P1371Q and heterozygous genotype for R30Q. While analysis of G1066G [32], and E514Q and P979L (this study; data not shown) showed no association with IBD with individual SNPs; a significant interaction was observed between P1371Q with P979L (0.002), G1066G (p = 0.004), R30Q (p = 0.005) respectively, with weak R30Q interactions with E514Q and P979L.
| Table 1: Association of DLG5 P1371Q with IBD in familial and sporadic IBD. | |||||
| In case-control | |||||
| Genotype | Control (n) | IBD (n) | OR | 95% CI | P v alue |
| CA | 10 | 27 | 2.335 | 1.097–4.972 | 0.0246 |
| CC | 160 | 185 | |||
| In the familial IBD registry | |||||
| Genotype | IBD (n) | All members (n) | OR | 95% CI | P value |
| CA | 10 | 23 | 1.665 | 0.771–3.597 | 0.1908 |
| CC | 148 | 193 | |||
| Table 2: Distribution of A allele of DLG5 SNPs P1371Q and R30Q in men and women. | |||||||||
| All | Males | Females | |||||||
| OR | 95% CI | P value | OR | 95%CI | P value | OR | 95%CI | P value | |
| R30Q | 2.131 | 1.233–3.684 | 0.0061 | 3.333 | 1.208–9.193 | 0.0158 | 1.525 | 0.751–3.098 | 0.2421 |
| P1371Q | 2.335 | 1.097–4.972 | 0.0246 | 1.141 | 0.383–3.400 | 0.8135 | 3.765 | 1.307–10.85 | 0.0095 |
| Table 3: Association of A allele of P1371Q and A allele of R30Q with IBD in a familial IBD registry and sporadic IBD. | |||||
| In case-control | |||||
| Genotype | Control (n) | IBD (n) | OR | 95% CI | P value |
| CA or GA | 32 | 73 | 2.265 | 1.405–3.652 | 0.0007 |
| Not (CA or GA) | 138 | 139 | |||
| C30m or G1371f | 10 | 45 | 4.311 | 2.101–8.846 | <0.0001 |
| Not (C30m or G1371f) | 160 | 167 | |||
| In the familial IBD registry | |||||
| Genotype | Control (n) | All members (n) | OR | 95% CI | P value |
| CA or GA | 32 | 72 | 1.805 | 1.125–2.897 | 0.0138 |
| Not (CC or GA) | 138 | 172 | |||
| C30m or G1371f | 10 | 37 | 2.860 | 1.380–5.925 | 0.0034 |
| Not (C30m or G1371f) | 160 | 207 | |||
This study confirmed that DLG5P1371Q was associated with IBD in both sporadic and familial IBD patients from a central Pennsylvania population. Gender distribution analysis showed that the A allele of P1371Q was significantly associated with IBD in women. Further study on association between P1371Q and R30Q showed an increased significance in disease association (OR = 2.265, 95% CI = 1.405–3.652, p = 0.0007). Analysis of the A allele of P1371Q in women with A allele of R30Q in men also showed an increased significance in the association with IBD (OR = 4.311, 95% CI = 2.101–8.846, p <0.0001). These results suggest that interaction between P1371Q and R30Q within DLG5is allelically complementary in susceptibility to IBD, with a dominant effect of R30Q.
DLG5 contains multiple functional domains distributed over the entire gene such as leucine zipper, PDZ1, PDZ2, SH3, and GUK domains [22]. Genetic variants in the DLG5 gene, especially in these domains may have impact on protein function. Phylogenetic analyses show that the DLG5 protein is more closely related to coiled-coil caspase recruitment domain (CARD) MAGUK proteins, which activate the NFkB pathway, than to the other DLG proteins. This indicates that DLG5 may function in pathways of host defense like other members of the CARD family (e.g. IBD-associated NOD2) [37]. G1066G is located at the second nucleotide of the 5’ end of exon 16 without changing the coding amino acid. In contrast, in silico analysis noted potential structural and functional implications of the variants R30Q and P1371Q [16]. The R30Q variant is located in the DUF622 domain and influences binding to Rab-GTPase, which is important in inflammatory signaling. P1371Q precedes the fourth DLG5 PDZ domain and is the third proline in a SH3 domain protein binding motif. This suggests that both variants could impair function of the DLG5 protein. Combined variants within DLG5 may result in increased impairment of protein function.
Our results indicate that the two synonymous SNPs, R30Q and P1371Q are significantly associated with IBD, while another DLG5 synonymous SNP G1066G is not associated with IBD. This suggests that the amino acid changes in these SNPs affect DLG5 protein function and thus play a role in IBD. We further investigated the epistatic interaction of the R30Q and P1371Q. Interactions may involve additive, synergistic, or antagonistic effects. Epistasis analysis from this study suggests that the interaction between R30Q and P1371Q is complementary. The evidence described above indicates that DLG5 plays a role in IBD susceptibility.
Recently, a growing body of evidence indicates that epistasis may play an important role in the formation and progression of human diseases [38, 39]. When epistasis occurs, the presence of two or more particular loci may increase or reduce the risk of a disease more than would be expected from their independent effects [40].
The detection and testing of epistasis requires powerful and modern statistical methods. More recently, a host of statistical models have been developed for analysing epistatic effects in different genetic designs including case-control studies [41, 42]. Among these, the recent model developed by R. Wu and his group has proven to be genetically meaningful through the incorporation of traditional quantitative genetic principles into statistical models [33]. Epistasis is partitioned into additive × additive, additive × dominant, dominant × additive and dominant × dominant components. Many studies can only estimate overall epistasis, but Wu’s quantitative model detects each of these components. Each component has physiologically significance. For example, additive × additive epistasis performs differently from dominant × dominant epistasis. If epistasis is due to the former, genotype AABB or aabb may display a different function from AAbb or aaBB. But if epistasis is due to the latter, AaBb should be different from other genotypes. With such knowledge, we can better choose an optimal treatment for IBD patients with different genotypes.
Our results show that R30Q displays a significant dominant × dominant genetic effect on the disease after adjusting for multiple tests by the conservative Bonferroni correction, while 1066G did not show any significance. This suggests that the combination between the heterozygote of R30Q and the heterozygote of P1371Q is significantly different for disease susceptibility from all other combinations. Significance levels were corrected for multiple comparisons, suggesting that our conclusions are statistically robust.
Recently, epistasis between TLR9 and IL23R and NOD2 [43], and the IL2/IL21 region and IL23R [44] has been reported. These results, together with those in this study, indicate that epistatic gene-gene interaction is an important component in IBD pathogenesis. To the best of our knowledge, the allelic complementation observed in P1371Q and R30Q is the first one that reports the detection of epistasis for IBD susceptibility between SNPs within a single IBD-associated gene. Further epistasis analysis will be valuable in elucidating the gene-gene interactions underlying IBD pathogenesis [45]. This will provide information for designing an experimental approach to confirm epistasis in animal models or human cell culture.
1 Russell RK, Nimmo ER, Satsangi J. Molecular genetics of Crohn’s disease. Curr Opin Genet Dev. 2004;14:264–70.
2 Schreiber S, Rosenstiel P, Albrecht M, Hampe J, Krawczak M. Genetics of Crohn disease, an archetypal inflammatory barrier disease. Nat Rev Genet. 2005;6:376–88.
3 Achkar JP, Duerr R. The expanding universe of inflammatory bowel disease genetics. Curr Opin Gastroenterol. 2008;24:429–34.
4 Henckaerts L, Figueroa C, Vermeire S, Sans M. The role of genetics in inflammatory bowel disease. Curr Drug Targets. 2008;9:361–8.
5 Ishihara S, Aziz MM, Yuki T, Kazumori H, Kinoshita Y. Inflammatory bowel disease: review from the aspect of genetics. J Gastroenterol. 2009;44:1097–108.
6 Haas SL, Abbatista M, Brade J, Singer MV, Bocker U. Interleukin-18 serum levels in inflammatory bowel diseases: correlation with disease activity and inflammatory markers. Swiss Med Wkly. 2009;139:140–5.
7 Yamazaki K, McGovern D, Ragoussis J, Paolucci M, Butler H, Jewell D, et al. Single nucleotide polymorphisms in TNFSF15 confer susceptibility to Crohn’s disease. Hum Mol Genet. 2005;14:3499–506.
8 Duerr RH, Taylor KD, Brant SR, Rioux JD, Silvernerg MJ, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314(5804):1461–3.
9 Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39:207–11.
10 Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39:596–604.
11 Libioulle C, Louis E, Hansoul S, Sandor C, Farnir F, Franchimont D, et al. Novel Crohn disease locus identified by genome-wide association maps to a gene desert on 5p13.1 and modulates expression of PTGER4. PLoS Genet. 2007;3:e58.
12 Wellcome Trust Case Control C. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–78.
13 Parkes M, Barrett JC, Prescott NJ, Tremelling M, Anderson CA, Fisher SA, et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nat Genet. 2007;39:830–2.
14 Latiano A, Palmieri O, Corritore G, Valvano MR, Bossa F, Cucchiara S, et al. Variants at the 3p21 locus influence susceptibility and phenotype both in adults and early-onset patients with inflammatory bowel disease. Inflamm Bowel Dis. 2009.
15 Lees CW, Satsangi J. Genetics of inflammatory bowel disease: implications for disease pathogenesis and natural history. Expert Rev Gastroenterol Hepatol. 2009;3:513–34.
16 Stoll M, Corneliussen B, Costello CM, Waetzig GH, Mellgard B, Koch WA, et al. Genetic variation in DLG5 is associated with inflammatory bowel disease. Nat Genet. 2004;36:476–80.
17 Wakabayashi M, Ito T, Mitsushima M, Aizawa S, Ueda K, Amachi T, Kioka N. Interaction of lp-dlg/KIAA0583, a membrane-associated guanylate kinase family protein, with vinexin and beta-catenin at sites of cell-cell contact. J Biol Chem. 2003;278:21709–14.
18 Humbert P, Russell S, Richardson H. Dlg, Scribble and Lgl in cell polarity, cell proliferation and cancer. Bioessays. 2003;25:542–53.
19 Daly MJ, Pearce AV, Farwell L, Fisher SA, Latiano A, Prescott NJ, et al. Association of DLG5 R30Q variant with inflammatory bowel disease. Eur J Hum Genet. 2005;13(7):835–9.
20 Newman WG, Gu X, Wintle RF, Liu X, van Oene M, Amos CI, Siminovitch KA. DLG5 variants contribute to Crohn disease risk in a Canadian population. Hum Mutat. 2006;27:353–8.
21 Friedrichs F, Brescianini S, Annese V, Latiano A, Berger K, Kugathasan S, et al. Evidence of transmission ratio distortion of DLG5 R30Q variant in general and implication of an association with Crohn disease in men. Hum Genet. 2006;119:305–11.
22 Friedrichs F, Stoll M. Role of discs large homolog 5. World J Gastroenterol. 2006;12:3651–6.
23 Vermeire S, Pierik M, Hlavaty T, Claessens G, van Schuerbeeck N, Joossens S, et al. Association of organic cation transporter risk haplotype with perianal penetrating Crohn’s disease but not with susceptibility to IBD. Gastroenterol. 2005;129:1845–53.
24 Lakatos PL, Fischer S, Claes K, Kovacs A, Molnar T, Altorjay I, et al. DLG5 R30Q is not associated with IBD in Hungarian IBD patients but predicts clinical response to steroids in Crohn’s disease. Inflamm Bowel Dis. 2006;12:362–8.
25 Ferraris A, Torres B, Knafelz D, Barabino A, Lionetti P, de Angelis GL, et al. Relationship between CARD15, SLC22A4/5, and DLG5 polymorphisms and early-onset inflammatory bowel diseases: an Italian multicentric study. Inflamm Bowel Dis. 2006;12:355–61.
26 Buning C, Geerdts L, Fiedler T, Gentz E, Pitre G, Reuter W, et al. DLG5 variants in inflammatory bowel disease. Am J Gastroenterol. 2006;101:786–92.
27 Torok HP, Glas J, Tonenchi L, Lohse P, Muller-Myhsok B, Limbersky O, et al. Polymorphisms in the DLG5 and OCTN cation transporter genes in Crohn’s disease. Gut. 2005;54:1421–7.
28 Tremelling M, Waller S, Bredin F, Greenfield S, Parkes M. Genetic variants in TNF-alpha but not DLG5 are associated with inflammatory bowel disease in a large United Kingdom cohort. Inflamm Bowel Dis. 2006;12:178–84.
29 Noble CL, Nimmo ER, Drummond H, Ho GT, Tenesa A, Smith L, et al. The contribution of OCTN1/2 variants within the IBD5 locus to disease susceptibility and severity in Crohn’s disease. Gastroenterol. 2005;129:1854–64.
30 Browning BL, Barclay ML, Bingham SA, et al. Gender-stratified analysis of DLG5 R30Q in 4707 Crohn’s disease patients and 4973 controls from 12 Caucasian cohorts. J Med Genet. 2008;45(1):36–42.
31 Browning BL, Huebner C, Petermann I, Demmers P, McCulloch A, Gearry RB, et al. Association of DLG5 variants with inflammatory bowel disease in the New Zealand Caucasian population and meta-analysis of the DLG5 R30Q variant. Inflamm Bowel Dis. 2007;13:1069–76.
32 Lin Z, Poritz L, Franke A, Li TY, Ruether A, Byrnes KA, et al. Genetic association of DLG5 R30Q with familial and sporadic inflammatory bowel disease in men. Dis Markers. 2009;27:193–201.
33 Wang Z, Liu T, Lin Z, Hegarty J, Koltun WA, Wu R. A general model for multilocus epistatic interactions in case-control studies. PLoS One 2010;5:e11384.
34 Wei Y, Zhenwu L, Ashley AK, John PH, Lisa SP, Yunhua W, et al. Association of a Nkx2-3 polymorphism with Crohn’s disease and expression of Nkx2-3 is up-regulated in B cell lines and intestinal tissues with Crohn’s disease 2009;3:189–95.
35 Lin Z, Cui X, Li H. Multiplex genotype determination at a large number of gene loci. Proc Natl Acad Sci. USA. 1996;93(6):2582–7.
36 Biank V, Friedrichs F, Babusukumar U, Wang T, Stoll M, Broeckel U, Kugathasan S. DLG5 R30Q variant is a female-specific protective factor in pediatric onset Crohn’s disease. Am J Gastroenterol. 2007;102:391–8.
37 Friedrichs F, Henckaerts L, Vermeire S, Kucharzik T, Seehafer T, Moller-Krull M, Bornberg-Bauer E, Stoll M, Weiner J, 3rd. The Crohn’s disease susceptibility gene DLG5 as a member of the CARD interaction network. J Mol Med. 2008;86:423–32.
38 Martin MP, Gao X, Lee JH, Nelson GW, Detels R, Goedert JJ, et al. Epistatic interaction between KIR3DS1 and HLA-B delays the progression to AIDS. Nat Genet. 2002;31:429–34.
39 Gabutero E, Moore C, Mallal S, Stewart G, Williamson P. Interaction between allelic variation in IL12B and CCR5 affects the development of AIDS: IL12B/CCR5 interaction and HIV/AIDS. AIDS. 2007;21:65–9.
40 Moore JH, Williams SM. Epistasis and its implications for personal genetics. Am J Hum Genet. 2009;85:309–20.
41 Zhang Y, Liu JS. Bayesian inference of epistatic interactions in case-control studies. Nat Genet. 2007;39:1167–73.
42 Gayan J, Gonzalez-Perez A, Bermudo F, Saez ME, Royo JL, Quintas A, et al. A method for detecting epistasis in genome-wide studies using case-control multi-locus association analysis. BMC Genomics. 2008;9:360.
43 Torok HP, Glas J, Endres I, Tonenchi L, Teshome MY, Wetzke M, et al. Epistasis between Toll-like receptor-9 polymorphisms and variants in NOD2 and IL23R modulates susceptibility to Crohn’s disease. Am J Gastroenterol. 2009;104:1723–33.
44 Glas J, Stallhofer J, Ripke S, Wetzke M, Pfennig S, Klein W, et al. Novel genetic risk markers for ulcerative colitis in the IL2/IL21 region are in epistasis with IL23R and suggest a common genetic background for ulcerative colitis and celiac disease. Am J Gastroenterol. 2009;104:1737–44.
45 Achkar JP, Fiocchi C. Gene-gene interactions in inflammatory bowel disease: biological and clinical implications. Am J Gastroenterol. 2009;104:1734–6.
Funding / potential competing interests: This work was supported by a grant from the Philadelphia Health Care Trust (to WA Koltun) and a Surgery Initiation Grant from Pennsylvania State University College of Medicine (to Z Lin).
Authors’ contributions: Z Lin and WA Koltun designed the research; Z Lin and JP Hegarty performed the experiments; A Berg, Z Wang and R Wu performed statistical analysis of the data, AA Kelly and Y Wang provided technical assistance; Z Lin, JP Hegarty, LS Poritz, and WA Koltun wrote the paper. The authors thank Tony Lin for his assistant with RFLP genotyping.