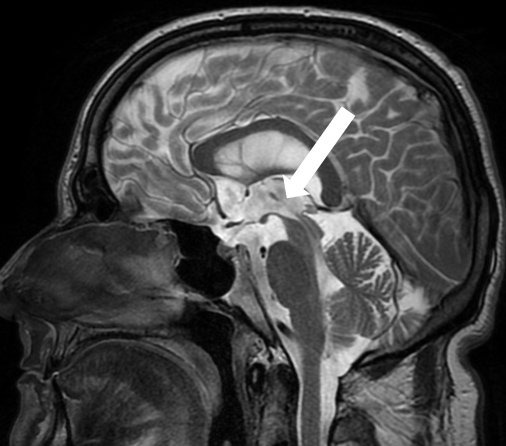
Figure 1
Brain MRI (T2) in sagittal plane of a patient with progressive supranuclear palsy (PSP). Note the marked midbrain atrophy adopting the appearance of a “hummingbird” (arrow).
DOI: https://doi.org/10.4414/smw.2011.13293
Parkinsonism is a neurological syndrome with four cardinal signs: bradykinesia, muscle rigidity, tremor at rest and impairment of postural reflexes. The diagnosis depends on the presence of bradykinesia, which denotes slowness of movements with a characteristic decrement on repetition (e.g. fatigue on finger tapping). Related symptoms are deterioration of handwriting (micrographia), slowing of walking (including, for instance, difficulty exiting a car), loss of facial expression (hypomimia) and speech volume (hypophonia). A key feature of bradykinesia is the difficulty of performing automatic sequential and simultaneous movements, which is essential for habitual motor control in activities of daily living (e.g. to get up and start walking, or walking while talking). Dopamine depletion of nigrostriatal pathways, ultimately leading to underactivation of basal ganglia thalamo-cortical loops, reflects the pathophysiological basis [1]. Recently, the motor impairment of bradykinesia has been conceptualised within the frame of a dual behavioural control system: a habitual (automatic) and a goal-directed system [2]. The former is characterised by rapid and parallel processing and the latter by slow and serial processing. Accordingly, in the parkinsonian state patients, due to the disrupted habitual loop, depend significantly more on activation of the relatively intact goal-directed system. Thus, usually highly automated movements such as walking lose their implicitness and become goal-directed, thereby requiring substantial attention resources. As a consequence, parkinsonian patients are “trapped” in an arduous goal-directed motor control [2].
Another cardinal motor manifestation of parkinsonism is the muscle stiffness called rigidity. It denotes the feeling of resistance when passively moving the patient’s limb, which is present throughout the range of motion. In milder cases it can be brought out by co-activation of the contralateral limb. Rigidity is less disabling than bradykinesia, but may enhance musculoskeletal pain associated with abnormal postures. A more socially than physically disabling feature of parkinsonism is the tremor at rest with its typical frequency of 5–6 Hz. It often involves the thumb and index finger, producing the stereotypic “pill-rolling” appearance. Resting tremor stops with action, but typically re-emerges within a few seconds when keeping the arms outstretched. When combined with rigidity it gives rise to the so-called “cogwheel” rigidity, the feeling of giving away in little jumps on examination. In contrast to bradykinesia, the pathophysiology of both rigidity and tremor is not well understood [3]. For resting tremor it has recently been suggested that the cerebello-thalamo-cortical loops are implicated rather than those involving the basal ganglia [4].
Finally, an important feature of parkinsonism is postural instability, often associated with gait disturbance. The main risk is falling. Hence the patient with parkinsonism should always be tested for postural instability, commonly performed by the “pull test”. In this test the patients are asked to maintain their balance while the examiner, standing behind, pulls forcefully backwards on their shoulders. The test is abnormal when the patient needs more than 2 steps to recover unaided (called retropulsion) or needs the examiner’s aid to prevent falling. Another risk of falling frequently encountered in parkinsonism is freezing of gait (FOG), denoting sudden motor blocks during walking, experienced by the patient as if glued to the floor. Narrow spaces (doors, bathrooms), but also set-shifting (start walking after standing up) or simultaneous movements (turning while walking) typically provoke FOG. The pathophysiology is probably linked to the mechanism underlying bradykinesia as discussed above with impaired automaticity (habitual control) representing the core deficit [5].
Parkinsonism can be classified into a broad spectrum of primary and secondary causes (table 1). Primary parkinsonism embraces neurodegenerative disorders of unknown or genetic origin. The most common type of primary parkinsonism is Parkinson’s disease (PD), an idiopathic disorder which is apparently sporadic in the majority of cases. It is estimated that about 10% of PD is familial, based on single gene mutations [6]. However, recent progress in genetic analysis, notably the genome-wide association studies (GWAS) [7], has made it possible to identify an increasing number of genetic susceptibility factors that may currently account for up to half of the risk of developing Parkinson’s disease [8]. For reasons that are outlined in the next section, it is clinically important and challenging to distinguish PD from the group of degenerative disorders commonly labelled as atypical parkinsonism. The designation “atypical” refers to the poor levodopa response of parkinsonism and the early manifestation of additional clinical features (Parkinson plus) such as ophthalmoparesis, dysautonomia, apraxia or dementia. The differential diagnosis of parkinsonism also encompasses a wide range of rare heredodegenerative disorders that are important to take into account if the clinical presentation is not typical, but this category is not further discussed here. Finally, secondary causes must always be ruled out as they are potentially treatable and common. In particular, drug-induced parkinsonism is prevalent and may be under-recognised in elderly patients [9].
| Table 1: Classification of parkinsonism. |
| Primary (degenerative) parkinsonism |
| – Parkinson’s disease (sporadic and genetic) |
| – Atypical parkinsonism (progressive supranuclear palsy, multisystem atrophy, corticobasal degeneration, Lewy body disease) |
| – Heredo-degenerative parkinsonisms (Wilson’s disease, GM1 Gangliosidosis etc.) |
| Secondary (acquired, symptomatic) parkinsonism |
| – Drugs (neuroleptics, antiemetics, antivertigo drugs) |
| – Vascular multiinfarct syndrome |
| – Toxins (manganese, CO, pesticides) |
| – Infectious (postencephalitic, neurosyphilis, HIV) |
| – Trauma (e.g., pugilistic encephalopathy) |
A major confounder in the early clinical diagnosis of PD is atypical parkinsonism. A clinico-pathological study has shown that a diagnostic accuracy of over 90% can be achieved by strict application of clinical criteria, but only at the cost of missing 32% of true PD cases [10]. Differentiation of PD and atypical parkinsonism is important, as the response to dopaminergic treatment is excellent in PD, but only poor or rapidly weaning in atypical parkinsonism [11]. Dopaminergic drugs may even be harmful by potentially worsening atypical features such orthostatic hypotension or confusion in cognitively impaired patients [12]. Also, patients with atypical parkinsonism should not be treated by deep brain stimulation (DBS), which is an effective treatment for PD only [13]. Finally, prognosis is better in PD, whereas atypical parkinsonism is generally associated with more rapid progression [14–17].
The most frequent atypical parkinsonian disorders are progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), multiple system atrophy (MSA) and dementia with Lewy bodies (LBD). PSP is characterised by symmetrical parkinsonism with supranuclear vertical gaze palsy, early falls (within the first year) and pseudobulbar palsy (dysarthria and dysphagia) as major parkinson-plus features [18]. Vertical gaze palsy affects predominantly downgaze, which can be easily overcome by vestibulo-ocular reflex (Doll’s manoeuvre), indicating that brainstem reflexes are relatively preserved until the later stages of the disease. Cognitive dysfunction is characterised by marked dysexecutive deficits while visuospatial abilities are relatively spared. Patients often appear disinhibited and unconcerned despite the severe physical disability. For instance, they jump up from the sitting position (“rocket sign”) or turn en bloc without considering the risk of falling associated with the postural instability. Recently, based on clinico-pathological findings, it has been suggested that a second form of PSP be distinguished, so-called PSP-Parkinsonism (PSP-P), and that the term Richardson syndrome (RS) be used for the typical disorder [19]. At least in the early stages PSP-P more closely resembles PD, with asymmetric onset and moderate response to dopaminergic treatment.
The classical description of CBD refers to an asymmetric form of parkinsonism combined with apraxia [20], which variably includes other basal ganglionic (e.g., dystonia) or cortical signs (alien limb phenomenon, tactile agnosia, myoclonus). Apraxia in CBD is typified by gestural deficits traditionally labelled as ideomotor apraxia, as well as impaired control of precise and individual finger movements, a hallmark of limb kinetic apraxia [21, 22]. Apraxia may be difficult to separate from dystonia or bradykinesia, and therefore distinguishable only on the less affected side. Apraxic deficits may be also present in PD, particularly of the limb kinetic type, but rather in the later stages of the disease and usually to a milder degree [23]. There is considerable overlap between PSP and CBD, with respect to both clinical and pathological features, giving rise to a revised nomenclature as discussed below in more detail.
In MSA the parkinsonism is combined with early autonomic failure and cerebellar deficits. Depending on the predominant symptom, a parkinsonian (MSA-P) and a cerebellar variant (MSA-C) are differentiated [24]. Autonomic deficits include orthostatic hypotension, bladder and erectile dysfunction. The latter may precede the onset of parkinsonism for years. Also, abnormal postures (PISA syndrome, anterocollis) or respiratory dysfunction, such as inspiratory stridor and/or sighs, are considered red flags that help to differentiate MSA from other atypical parkinsonian disorders [25]. Finally, in MSA the patient’s cognition is typically intact, often appearing in striking contrast to the extent of their physical disability.
Parkinsonism arising with early dementia is a core feature of DLB, which is likely in the spectrum of PD dementia (PDD). To delineate DLB from PDD, the somewhat arbitrary 1-year rule has been advocated, i.e. the emergence of a prominent cognitive decline within 12 months of disease onset [26]. Essential for the diagnosis of DLB is the presence of additional neuropsychiatric features such marked diurnal fluctuations in cognition and unprovoked visual hallucinations (often with children or hunting scenes). DLB should not be confused with diffuse Lewy body disease, which is a rapidly progressive dementia leading to death within 18 months, and considered rather in the differential diagnosis of Creutzfeld Jakob Disease [27].
Early postural instability, although most prominent in PSP, is common to all subtypes of atypical parkinsonism and therefore reflects an important red flag in the differential diagnosis of PD. Beyond the pull test, 10 steps of tandem gait may be a sensitive parameter in assessing postural instability and reliably differentiating PD from atypical parkinsonism within the first three years of disease onset [28]. In this study 92% of the PD patients showed intact performance (without side steps), which was possible only in 18% of the atypical cases. In contrast to what might have been expected, the discriminative value is even stronger with advancing age. Furthermore, a simple question on cycling ability may be a distinguishing factor, as more than half of atypical patients had stopped cycling 2½ years after disease onset, while few did in the PD group (“bicycle sign”) [29].
Oculomotor signs significantly contribute to discriminating between the various phenotypes of parkinsonism. Some features that may be differentiated at the bedside are summarised in table 2. As pointed out above, supranuclear gaze palsy, serving as its eponym, is the main characteristic of PSP, particularly RS. It is generally not a feature of PD and MSA, but may develop in late CBD [30] and in DLB [31]. Supranuclear gaze palsy in PSP initially affects downgaze and later involves upward gaze and horizontal planes as well. Slowing of vertical saccades may be an early sign of the disorder, which can be clinically appreciated by the loss of quick phases when testing vertical optokinetic nystagmus. In some patients in whom the range of ocular movements are still preserved, the so-called “round the houses” sign can be observed early [32], denoting a laterally curved trajectory of vertical saccades. Saccadic velocity may be slowed in CBD, however, usually in the later stages of the disease [33]. Saccadic velocity is not always easy to detect at the bedside, and therefore oculography (main sequence analysis) using infrared video recording may be a sensitive measure to reliably assess the speed of saccades [34]. Interestingly, it has recently been demonstrated that infrared video oculography could be useful in distinguishing PSP-P from PD [35], which is difficult on clinical grounds only. To interpret saccadic velocity in bedside testing the following rule can be helpful: “if you see the eyes move during a saccade, the saccades are too slow”. Saccadic latency in the horizontal plane is the most appropriate oculomotor feature to distinguish CBD early from other parkinsonian disorders. This delay in saccadic initiation may be referred to as “oculomotor apraxia”, which is prominent ipsilaterally to the more affected limb and has actually been shown to correlate with limb apraxia [30]. It is probably explained by the parietal atrophy of CBD. Impaired saccade triggering has also been found in LBD [36]. Square wave jerks (SWJ) are saccadic intrusions on steady gaze fixation, which are typically prominent early in PSP-RS and are thought to result from disinhibition of saccadic generators in brainstem [34]. To quantify an abnormal increase is difficult; however, more than 20 SWJs per minute are probably abnormal [37]. Increased SWJ’s may occur in the later stages of PD or CBD, and may be moderately present in MSA [38, 39]. There is no literature on LBD and SWJs; however, the prevalence is probably similar to that observed in late PD. Finally, impaired suppression of vestibulo-ocular reflex is frequent in MSA and may also be seen in PSP, thereby helping to differentiate other parkinsonian syndromes [38]. In addition to the oculomotor abnormalities, in PSP contracture of frontalis muscles with upraised eyebrows, conveying the characteristic stare, as well as eyelid disorders such as blepharospasms and eyelid apraxia, are fairly common. Furthermore, while glabellar sign (inability to suppress the blink on frontal finger tap) is generally seen in parkinsonism, its visual variant (elicited by bright light), along with photophobia, is more common in PSP [40].
Careful searching for atypical features in history and clinical examination is essential, as the frequently quoted levodopa challenge to a single dose is unreliable in early Parkinson’s disease. Furthermore, patients with atypical parkinsonism may also temporarily respond quite well to levodopa, particularly MSA-P and PSP-P. It is important to emphasise that autonomic dysfunction, postural instability, freezing or cognitive deficits are only atypical features if emerging early in the course of the disease. In the later stages these difficulties are also prevalent in PD. They represent a major source of disability frequently requiring nursing home placement [41]. Similarly, their diagnostic value for the subclassification of atypical parkinsonian disorders is restricted to earlier and middle stages. In advanced disease, when multiple systems become affected, differentiation is usually less consistent. For instance, oculomotor disturbances, emerging early in PSP, can be observed in late CBD as pointed out above, and conversely, apraxia defining CBD is common in advanced PSP [42].
| Table 2: Oculomotor signs in parkinsonism. | |||||
| PD | PSP | CBD | MSA | DLB | |
| Supranuclear gaze palsy | None | Prominent early (RS) | Possible, late | None | Possible |
| Saccades | Hypometric* | Slowed, early | Delayed**, early | Hypometric* | Triggering*, abnormal |
| Square wave jerks | Possible, late | Prominent,early | Possible, late | Moderate | Probably, late |
| PD = Parkinson’s disease, PSP = progressive supranuclear palsy, CBD = corticobasal degeneration, MSA = multiple system atrophy, DLB = dementia with Lewy bodies, RS = Richardson syndrome, MSA-C = cerebellar variant, *may be slowed late, ** may be slowed horizontally. | |||||
Differentiating primary from secondary causes of parkinsonism relies chiefly on taking of a detailed medical history and careful clinical examination. The most common secondary causes are vascular white matter disease (pseudoparkinsonism or lower body parkinsonism), which usually manifests with gait problems including freezing of gait with absent or only mild parkinsonian signs of the upper extremities and drug-induced parkinsonism, which should be suspected in patients treated with dopamine antagonists. Other toxic causes (e.g. manganese) as well as Wilson’s disease are uncommon but also important to recognise because they can be reversible or treatable. Since secondary parkinsonism is not the focus of this review, we will not further elaborate on it. In the vast majority of cases secondary causes can be ruled out clinically and with knowledge of the natural history of PD. Whenever doubts on the primary cause arise, either from the history, the clinical exam or the documented lack of levodopa response, further tests are indicated.
A new diagnostic challenge has recently emerged. Large drug trials in Parkinson’s disease and functional dopaminergic imaging ([18F]dopa PET, DaT SPECT) as surrogate markers of disease progression revealed that some 10% of the patients enrolled had normal dopaminergic functional imaging (scans without evidence of dopaminergic deficit or SWEDDs) [43–45]. The lack of dopaminergic deficit has been explained either by the low sensitivity of dopaminergic imaging techniques or by the absence of true parkinsonism. The former argument is highly unlikely, bearing in mind that reduced DaT density (reflecting the decline of striatal dopamine level) can already be observed in the preclinical stages. Furthermore, the first motor symptoms of Parkinson’s disease occur only after 80% of striatal and 50% of nigral dopamine cells are lost [46]. In addition, a clinical and electrophysiological study revealed that a significant proportion of the patients with “SWEDDs” had asymmetric rest tremor, but did not show the re-emergent type tremor typically seen in tremor-dominant PD [47]. Finally, true bradykinesia (with fatiguing) and hence parkinsonism was lacking. Instead, in SWEDDs subtle signs of limb dystonia, including head tremor, were present. Accordingly, increased cortical plasticity could be detected, a characteristic electrophysiological feature of dystonia which is reduced in PD. SWEDDs also had significantly fewer non-motor symptoms than PD patients, in particular normal smell identification tests [48]. These findings suggest that a large proportion of patients with SWEDDs have dystonic tremor, which can be difficult to distinguish from tremulous PD.
In recent years a number of clinico-pathological studies have been performed combining data-driven analysis of various clinical features with postmortem diagnosis using strict neuropathological criteria. These studies challenged the classification as outlined above by broadening the range of phenotypic variants within the traditional disease categories including PD. We are convinced that sorting out the subtypes of parkinsonian disorders, which increases the demand for expert counselling and management of patients, should have a place in clinical practice.
Traditionally, PD is subdivided in tremor-dominant and akinetic rigid subtypes, the former being considered more benign. In addition, a variant with rapid progression of postural instability and gait disturbance (PIGD), frequently seen in elderly patients, is commonly differentiated [49]. More recently, clinico-pathological studies using data-driven approaches, which are less biased by anecdotal views, extended the spectrum to 4 subtypes [50, 51]: earlier disease onset (EDO), tremor dominant (TD), non-tremor dominant (NTD) and rapid disease (RDP), [51] used motor symptoms in the first 5 years from disease onset for assignment of the patients to one of these subgroups, which is relevant since the motor phenotype often changes from tremor-dominant to non-tremor-dominant with disease progression [49]. It is therefore surprising that the non-tremulous phenotypes (69%) generally predominate. The study confirmed that the EOD subgroup had more motor fluctuations, a well-known complication of dopaminergic treatment in PD. However, the significantly shorter life expectancy (age of death 70.6 years) would not have been anticipated. In the early stages TD patients had lower disease severity, as expected, but the mean time to advanced disease was not different compared to NTD. In the NDT subgroup dementia was clearly more common (61%), along with more widespread cortical Lewy body pathology, than in EOD (16%) and TD (32%). Interestingly, the majority of the RDP subgroup (mean time to death: 7.7 years) started with tremor-dominant disease and more midline motor deficits. In conclusion, younger age of onset appears to be associated with more treatment-related motor complications, and a later age of onset with non-motor complications and more rapid motor progression. Furthermore, the notion of tremor as an independent indicator for slower disease progression [52] was not supported. The findings rather suggest, as pointed out above, that in cases of “benign tremulous PD”, an alternative diagnosis such as dystonia should be considered [46]. Finally, tremulous phenotypes generally appear to be less prevalent in early disease or may be even completely absent in the later stages [53].
The clinical syndrome of progressive supranuclear palsy was described in 1963 by Clifford Richardson, who highlighted the progressive disability due to postural instability and early falls, vertical supranuclear gaze palsy, mild cognitive changes, progressive axial rigidity and bulbar palsy. Pathologically, PSP is characterised by accumulation of tau protein and neuropil threads mainly in the pallidum, subthalamic nucleus, red nucleus, substantia nigra, oculomotor nucleus, medulla and dentate nucleus. Although characteristic, these pathological findings are not pathognomonic for PSP.
On the basis of their clinical presentation and distribution of tau pathology, several PSP phenotypes have been identified recently [54]. One clinico-pathological study of 103 pathologically proven PSP cases found two clusters of PSP phenotypes. 52% corresponded to the phenotype of the original description, with early falls, early cognitive dysfunction, abnormalities of gaze and postural instability (labelled “PSP-Richardson Syndrome” (PSP-RS)) [19]. 32% had a distinct clinical phenotype characterised by asymmetric onset, tremor, early bradykinesia, non-axial dystonia and a partial response to levodopa medications (labelled PSP-Parkinsonism (PSP-P)), which can be hard to differentiate from Parkinson’s disease in the early stages. PSP-RS had a significantly shorter disease duration compared to PSP-P (5.9 vs 9.1 years) and was pathologically characterised by a higher four-repeat/three-repeat tau ratio (2.84 vs 1.63) and by more severe tau pathology. 11% of these patients did not fit into one of these two clusters, suggesting a possible third PSP subtype not caught by this retrospective approach in which, for example, gait ignition failure was not extracted from the clinical notes. In a second study the same authors looked at patients from the Queen Square Brain Bank fulfilling clinical criteria for “pure akinesia with gait freezing” (PAGF) and found that this syndrome was infrequent but closely associated with PSP pathology, confirming PAGF as a third PSP subtype [55]. Apart from freezing of gait, axial rigidity and moderate akinesia, this phenotype was characterised by a longer disease duration (11 years), and less severe tau-accumulation compared to the classic phenotype. Furthermore, corticobasal syndrome (see below) or progressive non-fluent aphasia can be associated with PSP pathology [56]. Overall, the differences between these phenotypes are most prominent during the first two years and then tend to merge to a common clinical picture with severe postural instability, axial rigidity, bradykinesia and frontotemporal dementia. Characteristic eye-movement abnormalities usually occur later in PSP-P and PSP-PAGF, but may occasionally remain absent.
Recently the term corticobasal syndrome (CBS) has been introduced [57] as it is increasingly recognised that the clinical syndrome represented by CBD is frequently associated with other than CBD neuropathology [58, 59]. Conversely, in patients with histopathologically confirmed CBD only a minority showed the typical clinical picture with unilateral useless limb related to apraxia, dystonia and cortical sensory signs in the lifetime [59]. In fact, almost half of these patients presented with a PSP phenotype including early falls and supranuclear ophthalmoparesis. The authors therefore proposed to differentiate two major variants of CBD: CBD-CBS for the original syndrome as described by Rebeiz et al. [20], and CBD-Richardson’s syndrome to denote a subtype presenting with classical PSP features. In contradiction to these efforts at subclassification, it has been suggested that the PSP and CBD disorders be grouped together within the spectrum of frontotemporal dementias (so-called frontotemporal dementia/Pick complex) [60]. The rationale behind this view is that many of the patients with the primary movement disorders, if followed longitudinally, eventually develop frontotemporal behavioural and language difficulties, and vice versa. We generally acknowledge the clinical and pathological commonalities of many of neurodegenerative disorders and their final convergence to single multisystem disorders. However, we still favour a clinical approach that considers the subtypes of syndromes as being essential for the proper care and treatment of patients, at least in the early and middle stages of their disease.
Metabolic (SPECT, PET) or structural (MRI) imaging may improve the accuracy of clinical diagnosis for the various forms of parkinsonism. In particular, several SPECT tracers for dopamine transporter imaging (DaTSCAN) have become widely available and allow early differentiation of PD from other tremor syndromes (essential tremor, dystonia, SWEDDS) or vascular parkinsonism as discussed above [46]. However, DaTSCAN as a marker of presynaptic dopaminergic degeneration is not suitable to differentiate PD from atypical parkinsonism. Recently, FDG-PET analysis using sophisticated statistical methods reliably discriminates between PD, PSP and CBS, and may improve the diagnostic accuracy of PD by around 20% and several years before clinical diagnosis is certain [61]. The major disadvantage of PET is its high cost and the limited availability of the PET cyclotron.
Figure 1
Brain MRI (T2) in sagittal plane of a patient with progressive supranuclear palsy (PSP). Note the marked midbrain atrophy adopting the appearance of a “hummingbird” (arrow).
Routine structural MRI (T1 and T2) is not necessary in idiopathic PD responding well to levodopa, but is important in the differential diagnosis of atypical parkinsonism. It allows investigation of secondary causes (e.g. space-occupying lesions, normal pressure hydrocephalus etc.) as well as detection of structural alterations that are quite specific if present. For example, in PSP the midbrain atrophy may produce a characteristic appearance for which picturesque names have been introduced (e.g., “hummingbird sign”, fig. 1). In addition, in MSA-P a hyperintense MR signal at the outer border of the putamen may appear as a distinct “rim sign” [62] and the pontine atrophy in MSA-C may give rise to the “hot cross bun sign” [63]. However, these signs emerge rather late in the course of the disease. Further, on visual inspection they are subject to interpretation and have relatively low sensitivity. Recently, MRI imaging parameters have been developed that may allow differentiation of parkinsonism subtypes earlier and more reliably. In particular, brainstem MRI measures (pons-midbrain ratios and volume of superior cerebellar peduncle) may be important in discriminating PD, PSP and MSA [64, 65] as well as PSP subtypes [66]. In addition, severe cortical atrophy differentiates PSP-RS from PSP-P [67], paralleled by the severity of Tau pathology in brainstem and cortical areas. In future, automated analysis techniques of structural and functional imaging data may play a role in the diagnosis of neurodegenerative disorders [68], but hardly replace radiological skills of visual inspection completely.
Aggregation of misfolded proteins is considered a hallmark of the neurodegenerative process in parkinsonism. Therefore, parkinsonian disorders are conceived as proteinopathies involving two main types of protein: Tau and α-synuclein. Accordingly, proteinopathies are subdivided into Tauopathies and α-synucleinopathies. Tau is a microtubule-associated protein playing an important role in axonal transport. There are several isoforms comprising two main groups classified according to the repeat numbers in the microtubule-binding domain: 4 repeat (4R) and 3 repeat (3R) Tau. The former is prevalent in PSP, CBD and FTDP 17 and the latter in Pick’s disease. The PSP and CBS phenotype is highly specific for tauopathies [59]. α-Synuclein is a presynaptic protein whose function is unknown. Neuronal accumulation of α-synuclein defines PD and LBD while glial deposition is characteristic of MSA [69]. Hence this molecular pathological classification corresponds to specific phenotypic disorders and may influence clinical nosology in the future. Type and distribution of neuropathology determines clinical presentation [70]. For instance, 4R tauopathy predominant brainstem distribution is characteristic for PSP-P whereas predominant cortical deposition of α-synuclein containing Lewy bodies typifies LBD.
Levodopa remains the mainstay of treatment in Parkinson’s disease, although the armamentarium considerably widened with dopamine agonists, monoamine-oxidase (MAO-B), catechol-O-methyl transferase (COMT)-inhibitors and deep brain stimulation (DBS) introduced in recent decades. Dopamine agonists are considered first line therapy in patients aged up to 70, whereas levodopa is the first line treatment in patients aged over 70 with Parkinson’s disease. Levodopa also represents the treatment of choice in atypical parkinsonism since it is the most potent and best tolerated. In our view there is no need to use other medications that are generally less effective and have more side effects. Axial symptoms (dysphagia and postural instability), prevalent in atypical parkinsonism and little responsive to any dopaminergic treatment, may potentially be amenable to neurorehabilitation. Most studies on neurorehabilitative treatments have been done in PD, though [71]. For PSP, CBD and MSA mainly single case studies have been reported. The findings of a small controlled study suggested that complementation of balance rehabilitation by eye movement training improved stance and gait in PSP [72]. More research is clearly needed in the neurorehabilitative field. In general, patients with PD are at higher risk of osteoporosis, which must be considered and if necessary treated to avoid further complications [73].
In parkinsonism the differential diagnosis is broad. Identifying potentially treatable or even iatrogenic causes associated with secondary parkinsonism is obviously an important first step when encountering a patient. Levodopa response is a hallmark of idiopathic Parkinson’s disease and hence this diagnosis should not be missed either. It may be argued that differentiating atypical parkinsonisms has no merit, as specific treatments are not usually available. In addition, the clinico-pathological correlation is in any case variable and with disease progression the clinical symptoms considerably overlap, as reviewed above. Nonetheless, we strongly advocate a clinical approach based on a thorough history taking, neurological examination and ancillary investigations, which attempts to narrow down the specific syndrome associated with parkinsonism. By identifying a subtype of parkinsonism the prognosis can be estimated. For example, PSP-P is less disabling (e.g., no early falls), may respond temporarily to levodopa and shows a slower disease progression before eventually converting to full blown RS. Further, some of the atypical features such as autonomous or cognitive problems may be targeted by pharmacological treatment. Finally, and perhaps most importantly, identification of atypical parkinsonism may prevent unnecessary iatrogenic harm from ineffective drugs, to which these patients typically react very sensitively.
1 Graybiel AM. The basal ganglia. Curr Biol. 2000;10(14):R509–11.
2 Redgrave P, Rodriguez M, Smith Y, Rodriguez-Oroz MC, Lehericy S, Bergman H, et al. Goal-directed and habitual control in the basal ganglia: implications for Parkinson’s disease. Nat Rev Neurosci. 2010;11(11):760–72.
3 Hallett M. Parkinson revisited: pathophysiology of motor signs. Adv Neurol. 2003;91:19–28.
4 Benninger DH, Thees S, Kollias SS, Bassetti CL, Waldvogel D. Morphological differences in Parkinson’s disease with and without rest tremor. J Neurol. 2009;256(2):256–63.
5 Hallett M. The intrinsic and extrinsic aspects of freezing of gait. Mov Disord. 2008;23(Suppl 2):S439–43.
6 Gasser T. Molecular pathogenesis of Parkinson disease: insights from genetic studies. Expert Rev Mol Med. 2009;11:e22.
7 Wider C, Ross OA, Wszolek ZK. Genetics of Parkinson disease and essential tremor. Curr Opin Neurol. 2010;23(4):388–93.
8 Hardy J. Genetic analysis of pathways to Parkinson disease. Neuron. 2010;68 (2):201–6.
9 Esper CD, Factor SA. Failure of recognition of drug-induced parkinsonism in the elderly. Mov Disord. 2008;23(3):401–4.
10 Hughes AJ, Ben-Shlomo Y, Daniel SE, Lees AJ. What features improve the accuracy of clinical diagnosis in Parkinson’s disease: a clinicopathologic study. Neurology. 1992;42(6):1142–6.
11 Quinn N. Parkinsonism – recognition and differential diagnosis. BMJ. 1995;310(6977):447–52.
12 Jankovic J, Watts RL, Martin W, Boroojerdi B. Transdermal rotigotine: double-blind, placebo-controlled trial in Parkinson disease. Arch Neurol. 2007;64(5):676–82.
13 Shih LC, Tarsy D. Deep brain stimulation for the treatment of atypical parkinsonism. Mov Disord. 2007;22(15):2149–55.
14 Ben-Shlomo Y, Wenning GK, Tison F, Quinn NP. Survival of patients with pathologically proven multiple system atrophy: a meta-analysis. Neurology. 1997;48(2):384–93.
15 Jellinger KA, Wenning GK, Seppi K. Predictors of survival in dementia with lewy bodies and Parkinson dementia. Neurodegener Dis. 2007;4(6):428–30.
16 Nath U, Ben-Shlomo Y, Thomson RG, Lees AJ, Burn DJ. Clinical features and natural history of progressive supranuclear palsy: a clinical cohort study. Neurology. 2003;60(6):910–6.
17 Wenning GK, Litvan I, Jankovic J, Granata R, Mangone CA, McKee A, et al. Natural history and survival of 14 patients with corticobasal degeneration confirmed at postmortem examination. J Neurol Neurosurg Psychiatry. 1998;64(2):184–9.
18 Litvan I, Agid Y, Calne D, Campbell G, Dubois B, Duvoisin RC, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology. 1996;47(1):1–9.
19 Williams DR, de Silva R, Paviour DC, Pittman A, Watt HC, Kilford L, et al. Characteristics of two distinct clinical phenotypes in pathologically proven progressive supranuclear palsy: Richardson’s syndrome and PSP-parkinsonism. Brain. 2005;128(Pt 6):1247–58.
20 Rebeiz JJ, Kolodny EH, Richardson EP, Jr. Corticodentatonigral degeneration with neuronal achromasia. Arch Neurol. 1968;18(1):20–33.
21 Gebhardt A, Vanbellingen T, Baronti F, Kersten B, Bohlhalter S. Poor dopaminergic response of impaired dexterity in Parkinson’s disease: Bradykinesia or limb kinetic apraxia? Mov Disord. 2008;23(12):1701–6.
22 Leiguarda RC, Merello M, Nouzeilles MI, Balej J, Rivero A, Nogues M. Limb-kinetic apraxia in corticobasal degeneration: clinical and kinematic features. Mov Disord. 2003;18(1):49–59.
23 Vanbellingen T, Kersten B, Bellion M, Temperli P, Baronti F, Muri R, et al. Impaired finger dexterity in Parkinson’s disease is associated with praxis function. Brain and Cognition 2011; in press.
24 Wenning GK, Colosimo C, Geser F, Poewe W. Multiple system atrophy. Lancet Neurol. 2004;3(2):93–103.
25 Kollensperger M, Geser F, Seppi K, Stampfer-Kountchev M, Sawires M, Scherfler C, et al. Red flags for multiple system atrophy. Mov Disord. 2008;23(8):1093–9.
26 McKeith IG, Dickson DW, Lowe J, Emre M, O’Brien JT, Feldman H, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65(12):1863–72.
27 Gaig C, Valldeoriola F, Gelpi E, Ezquerra M, Llufriu S, Buongiorno M, et al. Rapidly progressive diffuse Lewy body disease. Mov Disord. 2011;26(7):1316–23.
28 Abdo WF, Borm GF, Munneke M, Verbeek MM, Esselink RA, Bloem BR. Ten steps to identify atypical parkinsonism. J Neurol Neurosurg Psychiatry. 2006;77(12):1367–9.
29 Aerts MB, Abdo WF, Bloem BR. The “bicycle sign” for atypical parkinsonism. Lancet. 2011;377(9760):125–6.
30 Vidailhet M, Rivaud S, Gouider-Khouja N, Pillon B, Bonnet AM, Gaymard B, et al. Eye movements in parkinsonian syndromes. Ann Neurol. 1994;35(4):420–6.
31 Brett FM, Henson C, Staunton H. Familial diffuse Lewy body disease, eye movement abnormalities, and distribution of pathology. Arch Neurol. 2002;59(3):464–7.
32 Quinn N. The “round the houses” sign in progressive supranuclear palsy. Ann Neurol. 1996;40(6):951.
33 Rivaud-Pechoux S, Vidailhet M, Gallouedec G, Litvan I, Gaymard B, Pierrot-Deseilligny C. Longitudinal ocular motor study in corticobasal degeneration and progressive supranuclear palsy. Neurology. 2000;54(5):1029–32.
34 Leigh RJ, Kennard C. Using saccades as a research tool in the clinical neurosciences. Brain. 2004;127(Pt 3):460–77.
35 Pinkhardt EH, Jurgens R, Becker W, Valdarno F, Ludolph AC, Kassubek J. Differential diagnostic value of eye movement recording in PSP-parkinsonism, Richardson’s syndrome, and idiopathic Parkinson’s disease. J Neurol. 2008;255(12):1916–25.
36 Mosimann UP, Muri RM, Burn DJ, Felblinger J, O’Brien JT, McKeith IG. Saccadic eye movement changes in Parkinson’s disease dementia and dementia with Lewy bodies. Brain. 2005;128(Pt 6):1267–76.
37 Abadi RV, Gowen E. Characteristics of saccadic intrusions. Vision Res. 2004;44(23):2675–90.
38 Anderson T, Luxon L, Quinn N, Daniel S, Marsden CD, Bronstein A. Oculomotor function in multiple system atrophy: clinical and laboratory features in 30 patients. Mov Disord. 2008;23(7):977–84.
39 Rascol O, Sabatini U, Simonetta-Moreau M, Montastruc JL, Rascol A, Clanet M. Square wave jerks in parkinsonian syndromes. J Neurol Neurosurg Psychiatry. 1991;54(7):599–602.
40 Kuniyoshi S, Riley DE, Zee DS, Reich SG, Whitney C, Leigh RJ. Distinguishing progressive supranuclear palsy from other forms of Parkinson’s disease: evaluation of new signs. Ann N Y Acad Sci. 2002;956:484–6.
41 Hely MA, Morris JG, Reid WG, Trafficante R. Sydney Multicenter Study of Parkinson’s disease: non-L-dopa-responsive problems dominate at 15 years. Mov Disord. 2005;20(2):190–9.
42 Zadikoff C, Lang AE, Klein C. The “essentials” of essential palatal tremor: a reappraisal of the nosology. Brain. 2006;129(Pt 4):832–40.
43 Dopamine transporter brain imaging to assess the effects of pramipexole vs levodopa on Parkinson disease progression. JAMA. 2002;287(13):1653–61.
44 Fahn S, Oakes D, Shoulson I, Kieburtz K, Rudolph A, Lang A, et al. Levodopa and the progression of Parkinson’s disease. N Engl J Med. 2004;351(24):2498–508.
45 Whone AL, Watts RL, Stoessl AJ, Davis M, Reske S, Nahmias C, et al. Slower progression of Parkinson’s disease with ropinirole versus levodopa: The REAL-PET study. Ann Neurol. 2003;54(1):93–101.
46 Kagi G, Bhatia KP, Tolosa E. The role of DAT-SPECT in movement disorders. J Neurol Neurosurg Psychiatry. 2010;81(1):5–12.
47 Schwingenschuh P, Ruge D, Edwards MJ, Terranova C, Katschnig P, Carrillo F, et al. Distinguishing SWEDDs patients with asymmetric resting tremor from Parkinson’s disease: a clinical and electrophysiological study. Mov Disord. 2010;25(5):560–9.
48 Silveira-Moriyama L, Schwingenschuh P, O’Donnell A, Schneider SA, Mir P, Carrillo F, et al. Olfaction in patients with suspected parkinsonism and scans without evidence of dopaminergic deficit (SWEDDs). J Neurol Neurosurg Psychiatry. 2009;80(7):744–8.
49 Alves G, Larsen JP, Emre M, Wentzel-Larsen T, Aarsland D. Changes in motor subtype and risk for incident dementia in Parkinson’s disease. Mov Disord. 2006;21(8):1123–30.
50 Lewis SJ, Foltynie T, Blackwell AD, Robbins TW, Owen AM, Barker RA. Heterogeneity of Parkinson’s disease in the early clinical stages using a data driven approach. J Neurol Neurosurg Psychiatry. 2005;76(3):343–8.
51 Selikhova M, Williams DR, Kempster PA, Holton JL, Revesz T, Lees AJ. A clinico-pathological study of subtypes in Parkinson’s disease. Brain. 2009;132(Pt 11):2947–57.
52 Josephs KA, Matsumoto JY, Ahlskog JE. Benign tremulous parkinsonism. Arch Neurol. 2006;63(3):354–7.
53 van Rooden SM, Colas F, Martinez-Martin P, Visser M, Verbaan D, Marinus J, et al. Clinical subtypes of Parkinson’s disease. Mov Disord. 2011;26(1):51–8.
54 Williams DR, Lees AJ. Progressive supranuclear palsy: clinicopathological concepts and diagnostic challenges. Lancet Neurol. 2009;8(3):270–9.
55 Williams DR, Holton JL, Strand K, Revesz T, Lees AJ. Pure akinesia with gait freezing: a third clinical phenotype of progressive supranuclear palsy. Mov Disord. 2007;22(15):2235–41.
56 Josephs KA, Boeve BF, Duffy JR, Smith GE, Knopman DS, Parisi JE, et al. Atypical progressive supranuclear palsy underlying progressive apraxia of speech and nonfluent aphasia. Neurocase. 2005;11(4):283–96.
57 Boeve BF, Lang AE, Litvan I. Corticobasal degeneration and its relationship to progressive supranuclear palsy and frontotemporal dementia. Ann Neurol. 2003;54(Suppl 5):S15–9.
58 Josephs KA, Petersen RC, Knopman DS, Boeve BF, Whitwell JL, Duffy JR, et al. Clinicopathologic analysis of frontotemporal and corticobasal degenerations and PSP. Neurology. 2006;66(1):41–8.
59 Ling H, O’Sullivan SS, Holton JL, Revesz T, Massey LA, Williams DR, et al. Does corticobasal degeneration exist? A clinicopathological re-evaluation. Brain. 2010;133(Pt 7):2045–57.
60 Kertesz A, McMonagle P, Blair M, Davidson W, Munoz DG. The evolution and pathology of frontotemporal dementia. Brain. 2005;128(Pt 9):1996–2005.
61 Tang CC, Poston KL, Eckert T, Feigin A, Frucht S, Gudesblatt M, et al. Differential diagnosis of parkinsonism: a metabolic imaging study using pattern analysis. Lancet Neurol. 2010;9(2):149–58.
62 Schrag A, Good CD, Miszkiel K, Morris HR, Mathias CJ, Lees AJ, et al. Differentiation of atypical parkinsonian syndromes with routine MRI. Neurology. 2000;54(3):697–702.
63 Drayer B, Burger P, Darwin R, Riederer S, Herfkens R, Johnson GA. MRI of brain iron. AJR Am J Roentgenol. 1986;147(1):103–10.
64 Paviour DC, Price SL, Jahanshahi M, Lees AJ, Fox NC. Regional brain volumes distinguish PSP, MSA-P, and PD: MRI-based clinico-radiological correlations. Mov Disord. 2006;21(7):989–96.
65 Quattrone A, Nicoletti G, Messina D, Fera F, Condino F, Pugliese P, et al. MR imaging index for differentiation of progressive supranuclear palsy from Parkinson disease and the Parkinson variant of multiple system atrophy. Radiology. 2008;246(1):214–21.
66 Longoni G, Agosta F, Kostic VS, Stojkovic T, Pagani E, Stosic-Opincal T, et al. MRI measurements of brainstem structures in patients with Richardson’s syndrome, progressive supranuclear palsy-parkinsonism, and Parkinson’s disease. Mov Disord. 2011;26(2):247–55.
67 Schofield EC, Hodges JR, Macdonald V, Cordato NJ, Kril JJ, Halliday GM. Cortical atrophy differentiates Richardson’s syndrome from the parkinsonian form of progressive supranuclear palsy. Mov Disord. 26(2):256–63.
68 Seeley WW, Crawford RK, Zhou J, Miller BL, Greicius MD. Neurodegenerative diseases target large-scale human brain networks. Neuron. 2009;62(1):42–52.
69 Wenning GK, Krismer F, Poewe W. New insights into atypical parkinsonism. Curr Opin Neurol. 2011.
70 Dickson DW, Ahmed Z, Algom AA, Tsuboi Y, Josephs KA. Neuropathology of variants of progressive supranuclear palsy. Curr Opin Neurol. 2010;23(4):394–400.
71 Ebersbach G, Ebersbach A, Edler D, Kaufhold O, Kusch M, Kupsch A, et al. Comparing exercise in Parkinson’s disease – the Berlin LSVT(R)BIG study. Mov Disord. 2010;25(12):1902–8.
72 Zampieri C, Di Fabio RP. Improvement of gaze control after balance and eye movement training in patients with progressive supranuclear palsy: a quasi-randomized controlled trial. Arch Phys Med Rehabil. 2009;90(2):263–70.
73 Gnädinger M, Mellinghoff HU, Kaelin-Lang A. Swiss Med Wkly. 2011 Feb 16;141:w13154. doi: 10.4414/smw.2011.13154.
Funding / potential competing interests: No financial support and no other potential conflict of interest relevant to this article was reported.