Is there a way to curb benzodiazepine addiction?
DOI: https://doi.org/10.4414/smw.2011.13277
AL
Lalive, U
Rudolph, C
Lüscher, KR
Tan
Summary
Benzodiazepines are widely prescribed drugs used to treat anxiety and insomnia, induce muscle relaxation, control epileptic seizures, promote anaesthesia or produce amnesia. Benzodiazepines are also abused for recreational purposes and the number of benzodiazepine abusers is unfortunately increasing. Within weeks of chronic use, tolerance to the pharmacological effects can develop and withdrawal becomes apparent once the drug is no longer available, which are both conditions indicative of benzodiazepine dependence. Diagnosis of addiction (i.e. compulsive use despite negative consequences) may follow in vulnerable individuals. Here, we review the historical and current use of benzodiazepines from their original synthesis, discovery and commercialisation to the recent identification of the molecular mechanism by which benzodiazepines induce addiction. These results have identified the mechanisms underlying the activation of midbrain dopamine neurons by benzodiazepines, and how these drugs can hijack the mesocorticolimbic reward system. Such knowledge calls for future developments of new receptor subtype specific benzodiazepines with a reduced addiction liability.
Introduction
The first benzodiazepine (BDZ) was synthesised in 1955 by Leo Sternbach [1] when he was working in Basel at Hoffmann–La Roche on the design of new tranquilisers. The project was initially abandoned because the pharmacological properties of the first compounds were disappointing. Two years later, one of the initially synthesised compounds was tested on animal behaviour. Subsequently, the compound showed very strong sedative, anticonvulsant and muscle relaxant effects in humans. These unexpected but impressive clinical findings led, in 1960, to the introduction of chlordiazepoxide into the clinical market under the brand name Librium®. A second compound, diazepam (Valium®), was subsequently marketed in 1963. For a while, these two molecules were the most successful sedative drugs on the market. The introduction of BDZs led to a significant decrease in the prescription of barbiturates [2], which are more toxic and have a higher risk of overdosing and an increased dependence liability. By the 1970s, BDZs had largely replaced the barbiturates previously used for sedation. Since then, nearly thirty different BDZs have been approved and marketed for clinical use.
BDZs enhance the effect of the neurotransmitter γ-aminobutyric acid (GABA), which results in five principal effects that have therapeutic benefits: sedation/hypnosis (for the treatment of insomnia), anxiolysis, control of epileptic seizures, myorelaxation and anterograde amnesia (as a premedication for invasive or otherwise unpleasant medical procedures) [3].BDZs are classified as either short-, intermediate- or long-acting. Although only subtle pharmacodynanic differences exist, short acting BDZs are preferred for the treatment of insomnia, while the longer-acting BDZs are recommended for the treatment of anxiety. In general, BDZs are safe and effective in the short term. Long-term use remains controversial because BDZs are prone to cause dependence and addiction [4], sometimes referred to as the sixth and unwanted effect [3].
BDZs are allosteric modulators of the GABAA receptors, which means that they only act in the presence of the endogenous ligand GABA. Based on efficacy, three groups of BDZs can be distinguished: the positive and the negative allosteric modulators, and the antagonists. The positive modulators potentiate GABAAR-evoked currents whereas the negative modulators decrease these currents. The antagonists prevent and reverse the effect of both types of allosteric modulators and have no consequences on the GABAAR-mediated currents on their own [3]. Only positive allosteric modulators such as chlordiazepoxide (Librium®) and diazepam (Valium®), and the antagonist flumazenil (Anexate®) are used therapeutically. All BDZs bind to a specific cleft located between the α and the γ subunit of the GABAA receptor [5]. Indeed, these ionotropic receptors are Cl- channels composed of five subunits with a specific stoichiometry of 2α, 2β and 1γ. Each subunit family exhibits several isoforms: α1−6, β1–3, γ1–3, and therefore the brain expresses a multitude of GABAA receptor combinations. Only α1-, α2-, α3- and α5-containing GABAA receptors are sensitive to BDZs, because they have a crucial histidine residue at homologous positions (α1H101, α2H101, α3H126 and α5H105) [6]. α4 and α6 subunit isoforms have an arginine residue at the homologous position, depriving α4-/α6- containing receptors of BDZ sensitivity [7, 8]. Based on these findings, transgenic knock-in (KI) mice were generated in which the histidine residue of the α subunit involved in the BDZ binding site was mutated to an arginine. As a consequence, GABAA receptors containing this specific α isoform become insensitive to BDZs while the physiological GABA-mediated inhibitory transmission remains intact in these mice. Studies using various KI mice allowed the identification of the role of the individual α isoforms in the six BDZ effects described above. GABAA receptors containing the α1 subunit mediate the sedative, the anterograde amnesic (partly), and the anticonvulsive effects of diazepam [9, 10]. Those containing the α2 isoform mediate the anxiolytic actions and to a large extent the myorelaxant effects [11, 12]. GABAA receptors containing the α3 or the α5 subunit isoforms also contribute to BDZ myorelaxant actions [12, 13], whereas GABAA receptors comprised of the α5 subunit isoform were shown to modulate the temporal and spatial memory effects of BDZs [13–15]. Recently, the addictive properties of BDZs have been shown to require the presence of α1-containing GABAA receptors [3, 16].
Addiction to benzodiazepines
BDZs were initially greeted with optimism by the medical profession, but, gradually, concerns arose. One major issue is the development of dependence and addiction symptoms after prolonged BDZ treatment [4, 17]. Dependence will occur in anybody who takes an addictive drug and is characterised by specific symptoms that are tolerance and withdrawal. Tolerance manifests when the patient must increase drug dosage in order to feel the same effects. Withdrawal symptoms are observed following discontinuation or abrupt reduction of BDZs dosage, even after a relatively short treatment period (three to four weeks). Such physiological symptoms are the main signs of physical dependence. The most frequent are insomnia, gastric problems, tremors, agitation, fearfulness and muscle spasms [18].Less frequently observed are irritability, sweating, depersonalisation, hypersensitivity to stimuli, depression, suicidal behaviour, psychosis, seizures and delirium tremens [19, 20]. Over-rapid withdrawal from BDZs also increases the severity of the symptoms. Slow and gradual reduction of dosage customised to the individual accompanied by psychological support are the most effective way of managing withdrawal [21]. Complete withdrawal can require four weeks to several years.
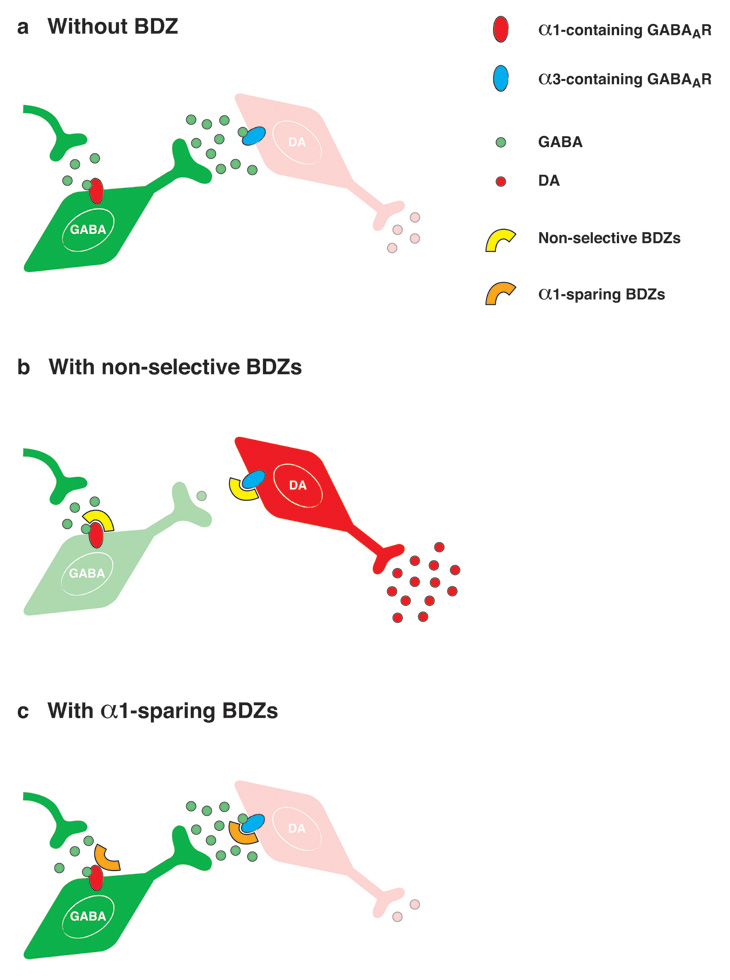
Figure 1
Mechanism for non-addictive benzodiazepines.
(a) The VTA is composed of three major cell types: GABA interneurons (green) and DA neurons (red). Glutamate neurons are not pictured for clarity purpose. Whereas interneurons express α1-containing GABAA receptors, DA neurons express GABAA receptors that contain other subunits, such as the α3 isoform. In the absence of BDZs, GABA neurons maintain a basal level of inhibition onto DA neurons.
(b) Clinically used BDZs such as diazepam (Valium®) bind to GABAA receptors and potentiate inhibitory GABAA evoked-currents on both cell types. However, the overall impact is much stronger in the interneurons because their GABAA receptors cause larger unitary currents than the one in DA neurons. Therefore interneurons are hyperpolarised and their activity is decreased. As a consequence, DA neurons are disinhibited and more DA is released, triggering early changes commonly induced by all addictive drugs.
(c) α1-sparing compounds bind to α1-containing GABAA receptors but antagonise the BDZ-binding site (misoriented symbol) and so do not affect the activity of GABA neurons. Hence DA neurons are not disinhibited, preventing an increase of DA release and consequent synaptic adaptations that may eventually lead to addiction.
If dependence can be diagnosed in most patients undergoing therapeutic BDZ treatment, the switch to an addicted state only happens in a fraction of drug users (20% for cocaine, less than 10% for BDZs [22]). Addiction is considered a brain disease defined by the World Health Organisation as compulsive substance intake despite negative consequences. It is also characterised by relapse after a prolonged period of abstinence, and so stands apart from dependence. Most often, people who recreationally abuse BDZs also abuse other drugs such as alcohol or opiods ( http://www.nida.nih.gov ). In that case, symptoms of dependence and addiction to BDZs manifest faster. For example, patients who have previously abused opioids abuse BDZs more frequently, at larger doses and for longer periods of time compared to BDZ only abusers [23]. However, little work has been done towards understanding cross modulation of pharmacological properties among drugs.
Genetic basis of addiction
As mentioned before not every drug consumer will suffer from addiction, and one hypothesis is that genetic predisposition underlies inter-individual vulnerabilities. Studies looking at identical and non-identical twin pairs revealed that when one twin was addicted to alcohol, the other identical twin had a high probability of getting addicted too. However, when a non-identical twin was addicted, the other twin did not necessarily develop an addiction. It was then proposed that addiction is partly due to genetic factors [24–26].Another study investigated the first-degree relatives (parents, siblings, or children) of 231 addicts and 61 non addicts. This analysis showed that if a parent had a drug or alcohol addiction, the child had an 8 times greater chance of developing an addiction [27]. Although genetic factors seem to be implicated, environmental influences should not be underestimated. So far there is no clear answer as to which are the sensible genes responsible for addiction, but specific gene mutations have been linked to sensitivity to alcohol and benzodiazepine consumption [28–30].
Another interesting question concerns the change in gene expression after drug abuse. Chronic psychostimulant use is well known to alter neuronal gene expression [31, 32]. Specifically, gene expression after a short cocaine treatment is dependent on CREB (cAMP response element-binding), a ubiquitously expressed protein, whereas gene expression after a long-term cocaine treatment is ΔFosB-dependent [33]. Both CREB and ΔFosB bind to specific DNA sequences, increasing or decreasing the transcription of downstream genes. ΔFosB accumulates in the nucleus accumbens (NAcc) and the striatum after repeated administration of drugs (cocaine, morphine). Several studies support the view that ΔFosB functions as a type of sustained “molecular switch” that gradually converts acute drug responses into relatively stable adaptations that contribute to the long-term neural and behavioural plasticity that underlies addiction [34, 35].
Recently it has been shown that psychostimulants also alter gene expression levels through epigenetic modifications, which means changes affecting the compaction of chromatin and thus DNA sequences’ accessibility to transcription factors. Two of the most studied epigenetic mechanisms are DNA methylation, that tightens chromatin, and histone acetylation, that loosens DNA packaging. In some cases these modifications of the transcriptome are heritable [36].
Cellular basis of addiction
Dependence and addiction symptoms are only observed after repeated drug exposure. However, a single injection is already sufficient to trigger synaptic adaptations in the brain that persist beyond elimination of the compound from the body [37, 38]. This so-called “drug-evoked synaptic plasticity” represents a permissive brain state that may, in the long term, lead towards the development of addiction in vulnerable subjects. Here we review where and how addictive drugs induce such lasting traces even after the first drug exposure.
The ventral tegmental area (VTA), at the origin of the mesolimbic system, consists of three neuronal subtypes: dopamine (DA) neurons, representing the majority of cells in the VTA (70%), γ-Aminobutyric acid (GABA) interneurons (15%, fig. 1a), and recently revealed glutamate neurons (15%) [39]. DA neurons release DA within the VTA and send projections to many different brain regions such as the NAcc, the striatum, the prefrontal cortex and hippocampus. These neurons are excited by unexpected natural rewards and inhibited by both absence of predicted rewards [40] and aversive stimuli [41]. Hence, the VTA is often referred to as the core nucleus of the reward system. GABA interneurons form synapses onto DA neurons and can control their activity by acting as a brake, and also project to the nucleus accumbens [42]. Glutamate cells send axons to the nucleus accumbens and the prefrontal cortex [39], but their role still remains elusive. Addictive drugs have been shown to hijack the mesolimbic reward system by disrupting normal functionality of this neuronal network.
A common particularity of addictive drugs is that they all target the VTA and acutely increase extracellular levels of DA within the VTA and target regions [43, 44]. This step happens within minutes after drug intake and is believed to be at the source of all forthcoming induction of synaptic plasticity and changes in network activity (see below) that happen either few hours after a single injection or after days of chronic drug use [45–47]. Drugs of abuse may therefore be classified by the three cellular mechanisms of action that have been described to cause this acute dopamine increase [48]. Group I includes opioids [49], cannabinoids [50] and γ-hydroxybutyrate (GHB) [51], which decrease the release of GABA from VTA interneurons and thereby remove the inhibitory transmission “brake” onto DA neurons. This indirect increase of DA cells’ activity is known as disinhibition, and is possible due to either cell-type specific expression of their respective receptor to the drug (as in opioids, cannabinoids), or higher affinity of the drug for the receptor located on GABA neurons (GHB). Nicotine constitutes Group II and directly activates DA neurons [52], whereas Group III drugs (including psychostimulants like cocaine and amphetamines) target and perturb the DA transporter (DAT) either by blocking it (cocaine) or reversing its activity (amphetamines) [53].
Neural basis for addictive properties of benzodiazepines
The pharmacological effects of BDZs depend crucially on the α subunit isoform identity. In the VTA, immunohistochemistry has revealed that the α1 subunit isoform is specifically expressed on GABA neurons (fig. 1). In corroboration, unitary currents recorded from GABA neurons are larger and slower than those recorded from DA neurons. The importance of this cellular difference was revealed when midazolam (MDZ, Dormicum®, a non-selective BDZ e.g., modulating all BDZ-sensitive GABAA receptors), a BDZ known to act as a positive modulator of GABAA receptors, was applied to the slice [3, 16]. The MDZ-induced potentiation of inhibition was stronger onto α1-expressing GABA neurons than onto DA neurons, which silenced the GABA neurons shutting down their transmitter release. As a consequence, BDZ lose their effect on DA neurons, simply because there is no GABA effect anymore to amplify, and the DA neurons are disinhibited (fig. 1b). In vivo, intravenous (iv) administration of MDZ led to a decrease in the firing rate of GABA cells and to an increase in DA neurons activity, thus in DA release. In the α1(H101R) KI mice, MDZ was unable to modulate inhibitory currents in GABA cells. Furthermore no change was observed in either GABA or DA neuron firing rate in vivo[3, 16]. These data indicate that BDZs can be classified as Group I addictive drugs and that they induce an indirect increase in extracellular DA levels by disinhibiting DA neurons (fig. 1b).
To investigate the consequences of the increase in extracellular DA levels, a common experiment is to inject a mouse intraperitoneally (ip) with a drug, slice the brain 24h later and assess synaptic transmission. Initially, it has been shown that after one shot of cocaine, the glutamatergic connection onto VTA DA neurons is strengthened [37]. Furthermore, this strengthening is blocked when the drug is co-administered with a DA D1-like receptor antagonist [46], or when given to a genetically modified mouse carrying a DA transporter insensitive to cocaine, with the result that the drug is unable to induce extracellular DA increase [47]. Since then, these observations have been repeated for all three classes of drugs, showing that this drug-evoked synaptic plasticity is DA-dependent and common to all addictive drugs [47, 54]. So far this strengthening of glutamate synapses represents the first known step in the chronology of events triggered by the initial DA increase after a single drug injection and implicated in the development of addiction disease. If there is no further exposure to the drug, the glutamatergic strengthening can go back to baseline after 7 days [37, 54]. However, if the animal is repetitively exposed to the drug, the synaptic plasticity extends to other regions of the brain [55] and other neurotransmitters, as GABAergic transmission onto DA neuron is decreased after 5 days of cocaine [55]. These changes in the mesolimbic network slowly add up and pave the way to addicted behaviour [55, 57].
Given the BDZs’ disinhibitory effect on DA neurons and the subsequent strong increase of DA in the target regions as well as in the VTA, it was then investigated if they could evoke any synaptic changes. Again, probing synaptic transmission in the VTA ex vivo and taking advantage of the development of the KI mice, plastic changes was observed by two groups independently [3, 16, 57]. 24 hours after either ip injection or direct intra-VTA delivery of MDZ, the typical drug-evoked synaptic plasticity was observed onto DA neurons. Coinjection of MDZ with flumazenil, a BDZ antagonist, blocked the plasticity. Interestingly, zolpidem (Stilnox®), an α1 selective non-classical BDZ available in clinics, evoked the plasticity. Conversely, in the α1(H101R)KI mice the glutamatergic connexion was unchanged after BDZ exposure. Furthermore, ip injection of L838,417 [10], an experimental BDZ, did not induce the plasticity. This compound modulates GABA-evoked currents generated by GABAA receptors expressing α2, α3 or α5 subunit isoforms. Importantly it is an antagonist at GABAA receptors containing the α1 isoform. This also argues for a specific role of the α1 subunit isoform in mediating addictive BDZ effects.
At a behavioural level, self-administration of addictive drugs, a paradigm that reveals reinforcing properties of a substance, has been shown in several animal models. Oral self-administration of MDZ was tested on WT and α1(H101R) KI mice. A free choice was given between a drinking bottle containing sucrose and another one containing both sucrose and MDZ. WT mice showed a clear preference for the MDZ-containing solution. However, α1(H101R) KI animals did not show any preference for the drug-containing solution, indicating that α1-containing GABAA receptors are required for the self-administration of MDZ [16]. Altogether, these data indicate that BDZs, like all other addictive drugs, are able to trigger adaptations in the reward system, such as increasing VTA DA cells activity and driving synaptic plasticity of excitatory inputs onto DA neurons. These adaptations depend on the specific expression of α1-containing GABAA receptors in GABA neurons of the VTA, mediating early changes induced by BDZs that may eventually lead to addiction.
Conclusions
BDZs continue to be widely used in medical practice for the treatment of anxiety and sleep disorders [59, 60] but are also increasingly popular for recreational purposes. However dependence and addiction are serious side effects, which cannot be neglected. Nevertheless cessation of BDZ prescription would be the waste of a valuable therapeutic tool. The results in animal models that we have reviewed here offer opportunities for the development of novel drugs addressing these side effects. It has been shown that α1-containing GABAA receptors mediate the addictive properties of BDZs. The α1-sparing compound L838,417 does not induce the changes in the brain that are observed with clinically used BDZs and other addictive drugs [16] and thus, L838,417 would be predicted to have no or at least a significantly reduced addictive potential compared to clinically used BDZs (fig. 1c). Unfortunately, the development of L838,417 was aborted due to unfavourable pharmacokinetic parameters in rodents [61]. Another fact to take in account is that α1-containing GABAAreceptors outside the VTA are presumably necessary for classical BDZs to induce sedative and anticonvulsant effects. Thus, α1-sparing compounds would be predicted not to exhibit these effects, but would still be efficient anxiolytics, an effect mediated by α2-containing GABAAreceptors. α1-sparing compounds might therefore represent non-sedating anxiolytics.
In any case, we believe that effort should be directed towards the development of α1-sparing compounds to render them safe and effective for future clinical use. The preclinical data reviewed here would suggest that such drugs provide many of the therapeutic benefits of BDZs, would likely be devoid of sedative side effects (which is actually useful when used for the treatment of anxiety disorders) and most importantly, lack addictive liability.
References
1 Sternbach LH. The benzodiazepine story. J Psychoactive Drugs. 1983;15:15–7.
2 Birdwood G, Craven M. Letter. Outmoded barbiturates. Br Med J. 1975;1:148.
3 Tan KR, Rudolph U, Lüscher C. Hooked on benzodiazepines: GABA(A) receptor subtypes and addiction. Trends in Neurosci. 2011;34:188–97.
4 Woods JH, Katz JL, Winger G. Benzodiazepines: use, abuse, and consequences. Pharmacol Rev. 1992;4:151–347.
5 Sigel E, Buhr A. The benzodiazepine binding site of GABAA receptors. Trends Pharmacol Sci. 1997;18:425–9.
6 Wieland HA, Lüddens H, Seeburg PH. A single histidine in GABAA receptors is essential for benzodiazepine agonist binding. J Biol Chem. 1992;267:1426–9.
7 Yang W, Drewe JA, Lan NC. Cloning and characterization of the human GABAA receptor alpha 4 subunit: identification of a unique diazepam-insensitive binding site. Eur J Pharmacol. 1995;291:319–25.
8 Korpi ER, Seeburg PH. Natural mutation of GABAA receptor alpha 6 subunit alters benzodiazepine affinity but not allosteric GABA effects. Eur J Pharmacol. 1993;247:23–7.
9 Rudolph U, Crestani F, Benke D, Brünig I, Benson JA, et al. Benzodiazepine actions mediated by specific gamma-aminobutyric acid(A) receptor subtypes. Nature. 1999;401:796–800.
10 McKernan RM, Rosahl TW, Reynolds DS, Sur C, Wafford KA, et al. Sedative but not anxiolytic properties of benzodiazepines are mediated by the GABA(A) receptor alpha1 subtype. Nat Neurosci. 2000;3:587–92.
11 Löw K, Crestani F, Keist R, Benke D, Brünig I, et al. Molecular and neuronal substrate for the selective attenuation of anxiety. Science. 2000;290:131–4.
12 Crestani F, Löw K, Keist R, Mandelli M, Möhler H, et al. Molecular targets for the myorelaxant action of diazepam. Mol Pharmacol. 2001;59:442–5.
13 Crestani F, Keist R, Fritschy J-M, Benke D, Vogt K, et al. Trace fear conditioning involves hippocampal alpha5 GABA(A) receptors. Proc Nat Acad Sci. 2002;99:8980–5.
14 Collinson N, Kuenzi FM, Jarolimek W, Maubach KA, Cothliff R, et al. Enhanced learning and memory and altered GABAergic synaptic transmission in mice lacking the alpha 5 subunit of the GABAA receptor. J Neurosci. 2002;22:5572–80.
15 Cheng VY, Martin LJ, Elliott EM, Kim JH, Mount HTJ, et al. Alpha5GABAA receptors mediate the amnestic but not sedative-hypnotic effects of the general anesthetic etomidate. J Neurosci. 2006;26:3713–20.
16 Tan KR, Brown M, Labouèbe G, Yvon C, Creton C, et al. Neural bases for addictive properties of benzodiazepines. Nature. 2010;463:769–74.
17 Salzman C. Addiction to benzodiazepines. The Psychiatric quarterly. 1998;69:251–61.
18 Pétursson H. The benzodiazepine withdrawal syndrome. Addiction. 1994;89:1455–9.
19 Gabbard GO. (2007) American Psychiatric Publishing 209-211.
20 http://en.wikipedia.org/wiki/Benzodiazepines - cite_note-isbn0-19-856667-0-85.
21 Colvin R. United States of America: Addicus Books (2008)74–76.
22 Wagner FA, Anthony JC. From first drug use to drug dependence; developmental periods of risk for dependence upon marijuana, cocaine, and alcohol. Neuropsychopharmacology. 2002;26:479–88.
23 Rooney S, Kelly G, Bamford L, Sloan D, O’Connor JJ. Co-abuse of opiates and benzodiazepines. Ir J Med Sc. 1999;168:1,36–41.
24 Prescott CA, Kendler KS. Genetic and environmental contributions to alcohol abuse and dependence in a population-based sample of male twins. Am J Psy. 1999;156:34–40.
25 Enoch MA. Goldman D. The genetics of alcoholism and alcohol abuse. Cur Psy Rep. 2001;3:144–51.
26 Goldman D, Oroszi G, Ducci F. The genetics of addictions: uncovering the genes. Nat Rev Gen. 2005;6:521–32.
27 Merikangas KR, Mehta RL, Molnar BE, Walters EE, Swendsen JD, et al. Comorbidity of substance use disorders with mood and anxiety disorders: results of the International Consortium in Psychiatric Epidemiology. Add Behav. 1998;23:893–907.
28 Hu X, Oroszi G, Chun J, Smith TL, Goldman D, et al. An expanded evaluation of the relationship of four alleles to the level of response to alcohol and the alcoholism risk. Alc Clin Exp Res. 2005;29:8–16.
29 Schuckit MA, Mazzanti C, Smith TL, Ahmed U, Radel M, et al. Selective genotyping for the role of 5-HT2A, 5-HT2C, and GABA alpha 6 receptors and the serotonin transporter in the level of response to alcohol: a pilot study. Biol Psy. 1999;45:647–51.
30 Iwata N, Cowley DS, Radel M, Roy-Byrne PP, Goldman D. Relationship between a GABAA alpha 6 Pro385Ser substitution and benzodiazepine sensitivity. Am J Psy. 1999;156:1447–9.
31 White FJ, Kalivas PW. Neuroadaptations involved in amphetamine and cocaine addiction. Drug Alc Dep. 1998;51:141–53.
32 Nestler EJ. Molecular neurobiology of addiction. Am J Psy. 2001;10:201–17.
33 McClung CA, Nestler EJ. Regulation of gene expression and cocaine reward by CREB and DeltaFosB. Nat Neurosci. 2003;6:1208–15.
34 McClung CA, Ulery PG, Perrotti LI, Zachariou V, Berton O, et al. DeltaFosB: a molecular switch for long-term adaptation in the brain. Brain Res Mol Brain Res. 2004;132:146–54.
35 Nestler EJ. Review. Transcriptional mechanisms of addiction: role of DeltaFosB. Biol Sci. 2008;363:3245–55.
36 Maze I, Nestler EJ. The epigenetic landscape of addiction. Annals New York Acad Sci. 2011;1216:99–113.
37 Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001;411:583–7.
38 Lüscher C, Malenka RC. Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron. 2011;69:650–63.
39 Yamaguchi T, Sheen W, Morales M. Glutamatergic neurons are present in the rat ventral tegmental area. Eur J Neurosci. 2007;25:106–18.
40 Schultz W, Dayan P, Montague PR. A neural substrate of prediction and reward. Science. 1997;275:1593–9.
41 Ungless MA, Magill PJ, Bolam JP. Uniform inhibition of dopamine neurons in the ventral tegmental area by aversive stimuli. Science. 2004;303:2040–2.
42 Xia Y, Driscoll JR, Wilbrecht L, Margolis EB, Fields HL, Hjelmstad GO. Nucleus accumbens medium spiny neurons target non-dopaminergic neurons in the ventral tegmental area. J Neurosci. 2011;31(21):7811-6.
43 Di Chiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Nat Acad Sci. 1988;85:5274–8.
44 Nestler EJ. Is there a common molecular pathway for addiction? Nat. Neurosci. 2005;8:1445–9.
45 Di Chiara G, Bassareo V, Fenu S, De Luca MA, Spina L, et al. Dopamine and drug addiction: the nucleus accumbens shell connection. Neuropharmacol. 2004;47(Suppl 1):227–41.
46 Argilli E, Sibley DR, Malenka RC, England PM, Bonci A. Mechanism and time course of cocaine-induced long-term potentiation in the ventral tegmental area. J Neurosci. 2008;28:9092–100.
47 Brown MTC, Bellone C, Mameli M, Labouèbe G, Bocklisch C, et al. Drug-Driven AMPA Receptor Redistribution Mimicked by Selective Dopamine Neuron Stimulation. PLoS ONE 2010;5: e15870.
48 Lüscher C, Ungless MA. The mechanistic classification of addictive drugs. PLoS Med. 2006;3:e437.
49 Johnson SW, North RA. Opioids excite dopamine neurons by hyperpolarization of local interneurons. J Neurosci. 1992;12:483–8.
50 Szabo B, Siemes S, Wallmichrath I. Inhibition of GABAergic neurotransmission in the ventral tegmental area by cannabinoids. Euro J Neurosci. 2002;15:2057–61.
51 Cruz HG, Ivanova T, Lunn M-L, Stoffel M, Slesinger PA, et al. Bi-directional effects of GABA(B) receptor agonists on the mesolimbic dopamine system. Nat Neurosci. 2004;7:153–9.
52 Maskos U, Molles BE, Pons S, Besson M, Guiard BP, et al. Nicotine reinforcement and cognition restored by targeted expression of nicotinic receptors. Nature. 2005;436:103–7.
53 Sulzer D, Sonders MS, Poulsen NW, Galli A. Mechanisms of neurotransmitter release by amphetamines: a review. Prog Neurobiol. 2005;75:406–33.
54 Saal D, Dong Y, Bonci A, Malenka RC. Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron. 2003;37:577–82.
55 Mameli M, Halbout B, Creton C, Engblom D, Parkitna JR, et al. Cocaine-evoked synaptic plasticity: persistence in the VTA triggers adaptations in the NAc. Nat Neurosci. 2009;12:1036–41.
56 Liu Q-, Pu L, Poo M. Repeated cocaine exposure in vivo facilitates LTP induction in midbrain dopamine neurons. Nature. 2005;437:1027–31.
57 Wolf ME. The Bermuda Triangle of cocaine-induced neuroadaptations. Trends Neurosci. 2010;33:391–8.
58 Heikkinen AE, Möykkynen TP, Korpi ER. Long-lasting modulation of glutamatergic transmission in VTA dopamine neurons after a single dose of benzodiazepine agonists. Neuropsychopharmacology. 2009;34:290–8.
59 Anderson KN, Shneerson JM. Drug treatment of REM sleep behavior disorder: the use of drug therapies other than clonazepam. J Clinic Sleep Med. 2009;5:235–9.
60 Hollister LE, Müller-Oerlinghausen B, Rickels K, Shader RI. Clinical uses of benzodiazepines. J Clinic Psychopharmacol. 1993;13:1S–169S.
61 Scott-Stevens P, Atack JR, Sohal B, Worboys P. Rodent pharmacokinetics and receptor occupancy of the GABAA receptor subtype selective benzodiazepine site ligand L-838417. Biopharma Drug Disp. 2005;26:13–20.