Low molecular weight hyaluronan, via AP-1 and NF-κB signalling, induces IL-8 in transformed bronchial epithelial cells
DOI: https://doi.org/10.4414/smw.2011.13255
CD
Ochoa, HG
Garg, CA
Hales, DA
Quinn
Summary
QUESTIONS UNDER STUDY: New evidence demonstrated that high tidal volume mechanical ventilation results in substantial bronchial airway mechanical strain. In addition, high tidal volume mechanical ventilation has been shown to increase IL-8 production in a mechanism mediated, at least in part, by low molecular weight hyaluronan (LWM-HA). In the present study, it was investigated whether LMW-HA synthesised in the lung, in response to cyclic stretch, increased IL-8 production in the bronchial epithelium.
METHODS: This question was approached by stimulating a transformed human bronchial epithelial cell line with LMW-HA isolated from stretched human lung fibroblasts and probed for the activation of extracellular signal-regulated kinase pathways.
RESULTS: LMW-HA increased IL-8 secretion in transformed bronchial epithelial cells. Additionally, LMW-HA augmented the levels of phospho c-Jun NH2-terminal kinase (JNK) and phospho extracellular signal-regulated kinase 1/2 (ERK1/2), and also mobilised nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) from the cytoplasm to the nucleus. The inhibition of JNK, ERK1/2 and NF-κB blocked IL-8 secretion in response to LMW-HA.
CONCLUSION: The data suggest that LMW-HA produced by lung fibroblasts in response to cyclic stretch increases the secretion of IL-8 in transformed bronchial epithelial cells via AP-1 and NF-κB signalling pathways. These findings support the hypothesis that LMW-HA plays an active role in acute lung inflammation triggered by mechanical strain.
List of abbreviations used
ANOVA: Analysis of Variance; AP-1: Activator Protein-1; BEGM: Bronchial Epithelial Cell Growth System; DNA: deoxyribonucleic acid; ELISA: Enzyme-linked Immunosorbent Assay; GAG: Glucosaminoglycans; HA: Hyaluronan; HAdase: Hyaluronydase; HAS: Hyaluronan Synthase; HMW-HA: High Molecular Weight Hyaluronan; IFN: Interferon; IL-8: Interleukin-8; JNK: c-Jun NH2-terminal Kinase; ERK 1/2: Extracellular Signal-regulated Kinase 1/2; JAK/STAT pathway: Janus Kinase/Signal Transducers and Activator of Transcription pathway; LDH: Lactate dehydrogenase; LMW-HA: Low Molecular Weight Hyaluronan; NF-κB: Nuclear Factor kappa-light-chain-enhancer of activated B cells; MAPK: Mitogen-activated Protein Kinase; TLR: Toll-like Receptors; TNF: Tumour Necrosis Factor; VALI: Ventilator-associated Lung Injury.
Introduction
Hyaluronan (HA) – first discovered in 1934 – is a non-sulphated linear glycosaminoglycan (GAG) composed of repeating disaccharides (β, 1-3 D-N-acetylglucosamine, β, 1-4 D-glucuronic acid) [1]. HA normally occurs as a part of the extracellular matrix in a high molecular weight (HMW) polymer form (>1000 kDa) [2, 3]. Low molecular weight HA (LMW, defined as <500 kDa) can be produced by the breakdown of HMW-HA to LMW-HA, or by de novosynthesis by hyaluronan synthases. Traditionally, it was believed that the physiological function of HA was only structural [4]. However, HA serves as a pro-inflammatory molecule. Recent studies revealed increased amounts of total HA and LMW-HA in animal models of acute lung injury [5–8]. Moreover, LMW-HA can induce the expression of several pro-inflammatory cytokines, including interleukin-8 (IL-8) [1, 9–12].
New evidence has demonstrated that high tidal volume mechanical ventilation results in substantial bronchial airway mechanical strain [13]. Our laboratory has investigated the role of HA in ventilator-associated lung injury (VALI) [1, 2, 4, 6]. Using in vitroand in vivomodels of VALI, we found that cyclic stretch produced LMW-HA though the janus kinase/signal transducers and activator of transcription pathway (JAK/STAT) signalling pathway, and that LMW-HA induced IL-8 expression. However, the downstream mechanism by which LMW-HA induces IL-8 expression in the lung is unclear. Moreover, whether LMW-HA plays a role in bronchial injury induced mechanical strain is unknown. To this end, we evaluated IL-8 production in human bronchial epithelial cells in response to LMW-HA synthesised by stretched human lung fibroblasts. In order to determine the response of IL-8 expression by HA size and species, we compared the results of LMW-HA synthesised by stretched human lung fibroblasts to LMW-HA from bovine vitreous and HMW-HA from rooster comb. It was found that newly synthesised LMW-HA increases IL-8 production via activator protein-1 (AP-1) and NF-κB signalling pathways.
Materials and methods
Maintenance of cells
SV40 transformed human bronchiolar epithelial cells, BEAS-2B cells, were used to test the pro-inflammatory effects of HA. BEAS-2B cells were maintained in BEGM bullet kit media (Clonetics Inc., San Diego, CA). Cells were seeded at 3 × 106cells/100 mm dish and incubated at 37 °C in a humidified atmosphere with 5% CO2 for 48 hrs prior to HA stimulation, with and without inhibitors. IMR-90 human foetal fibroblasts (Coriell Labs, Camden, NJ) were maintained in Rosewell Park Memorial Institute (RPMI) 1640 medium (Gibco, Grand Island, NY) supplemented with 20% foetal bovine serum (FBS - Gibco, Grand Island, NY), 1% penicillin/streptomycin and 0.1% amphotericinum B.
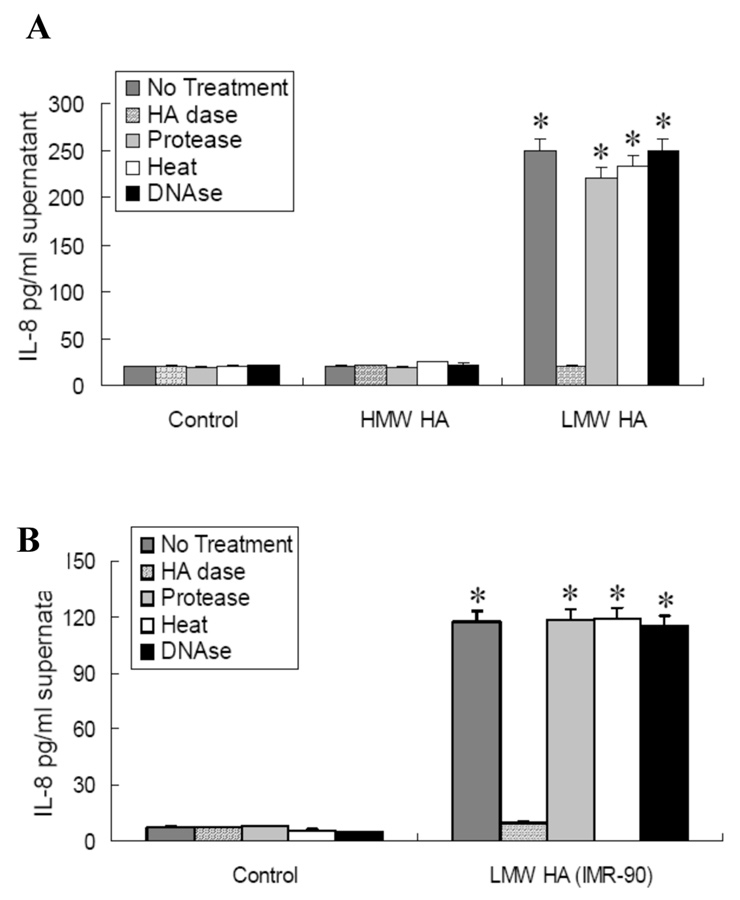
Figure 1
IL-8 production was stimulated by LMW-HA but not by HMW-HA in human transformed bronchial epithelial cells (BEAS-2B cells)
BEAS-2B cells were incubated with LMW-HA, HMW HA, or no HA in serum-free media for 6 hours.
A. Effect of LMW-HA from bovine vitreous humor and HMW HA from rooster comb on IL-8 production (n = 5);
B.Effect of LMW-HA isolated from IMR-90 fibroblasts (n = 3) on IL-8 production. HAdase treatment completely blocked LMW-HA-induced IL-8 production. Protease treatment, heat treatment and DNAse pre-treatment had no effect on LMW-HA-induced IL-8 production.
*p<0.05 vs HMW HA exposure and no HA control.
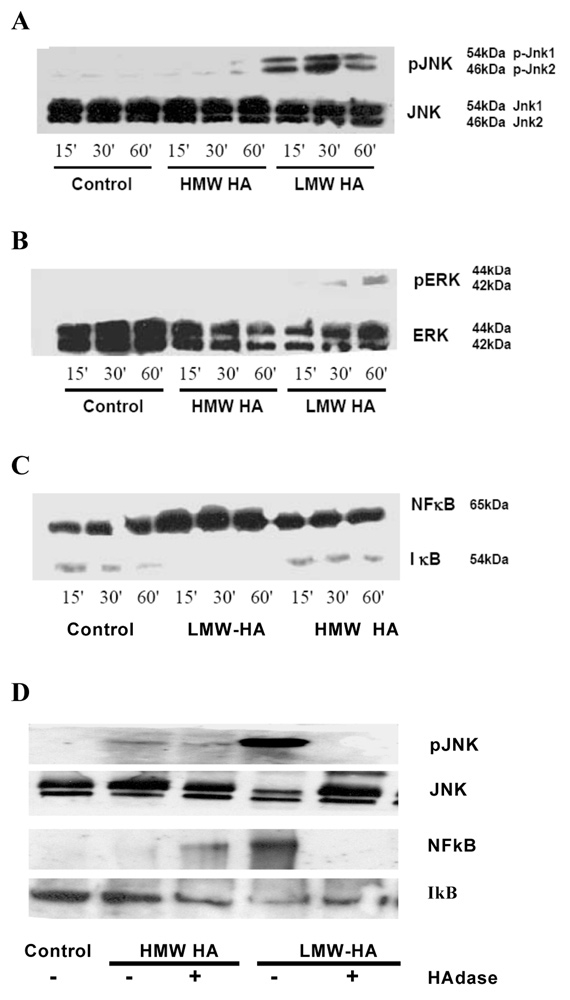
Figure 2
LMW-HA from bovine vitreous humor, but not HMW HA or HAdase treated LMW-HA from rooster comb, activated the JNK, ERK and NF-κB/IκB pathways in human BEAS-2B bronchial epithelial cells.
Western blot analysis of effects of LMW-HA on BEAS-2B human transformed bronchial epithelial cells:
A.c-Jun NH2-terminal kinase,
B.ERK1 and ERK-2, and
C.NF-κB in nuclear fraction / IκB in cytosol fraction,
D. c-jun N-terminal kinase, NF-κB and IκB expression with HAdase treated HMW and LMW-HA.
Each blot is representative of 3 separate experiments, and was quantified using densitometry. Results are expressed as fold-change compared with control in arbitrary units.
* p<0.05 vs control.
‡ p<0.05 vs HMW-HA
Mechanical stretch of lung fibroblasts
The IMR-90 cells were stimulated with cyclic stretch to produce LMW-HA, as previously described [6]. Fibroblasts were seeded at 2 × 106 cells/dish onto sterile silicone membranes mounted in a polypropylene 100 mm dish. This stretching device was custom built and provided by Martha Gray Ph.D (Massachusetts Institute of Technology, Cambridge, MA) [14]. This stretch apparatus provides a sinusoidal, spatially homogeneous, and isotropic biaxial strain to cultured cells. Cells were subjected to stretch in 37 °C and 5% CO2 at 60 cycles/minute with 15% strain for 4 hours. In addition, it was previously estimated that a change from 42% to 64% of total lung capacity corresponded to a 15% of linear strain [15]. This protocol was chosen because it induced maximal hyaluronan production and did not affect lactate dehydrogenase (LDH) release from stretched cells. LDH was measured in static cells supernatant vs. stretched cells supernatant as described by our group before [16].
Extraction and Analysis of HA
HA was extracted from pooled cell-free culture supernatants under dissociative conditions using 4 M guanidine HCl, as previously described ([6]. The size of the HAs used has been previously reported and were determined using column chromatography [6]. Briefly, conditioned media form stretched fibroblasts were lyophilised against water. HA presence was confirmed by proteoglycan electrophoresis as described earlier [1]. HA was purified using Sepharose CL-4B size exclusion chromatography (150 cm × 2 cm) equilibrated in 0.05 M sodium acetate buffer pH 6.7. Calibration of the column occurred as follows: void volume (Vo) was measured using Dextran blue 2000; total volume (Vt) was measured using uronic acid; HA sizes were determined by relative elution volume (Kaw) compared with three standards of known molecular weight (1600 kDa; 370 kDa; and 178 kDa). All samples were analysed by the same method and purified for further analysis. We also confirmed the purity of HA in the column fraction of stretched fibroblasts by FT-IR spectroscopy analysis. We have previously reported that the molecular weight of HA produced by stretched fibroblasts is <500 kDa with an average weight of 178 kDa [1].
Stimulation of BEAS-2B cells with HA
LMW-HA (370 kDa) from bovine vitreous humor was purchased from Sigma Chemical Co. (St. Louis, MO) and ICN Biochemicals (Costa Mesa, CA), and HMW-HA (1600 kDa) from rooster comb was purchased from Sigma Chemical Co (St. Louis, MO). Static cultures of BEAS-2B cells were washed with HBSS and then treated with LMW-HA, HMW-HA, or IMR-90 HA for 6 hours at 37 °C, at a concentration of 100 µg/ml in serum-free media. Possible contamination of HA samples by lipopolysaccharide (LPS) was controlled for by adding 10 µg/ml of polymyxin. LPS was quantified in cultures by a Limulus endotoxin assay (Sigma Chemical Co.). Less than 0.03 endotoxin units (EU)/ml (a negative) was detected in the HA samples. A dose of LMW-HA at 100 µg/ml was chosen to maximise IL-8 production. In a previous report, it was found that the maximum IL-8 production in alveolar type II-like cells was induced by LMW-HA at 100 µg/ml [1].
To rule out effects of contaminating proteins in HA samples, cells were treated with heat inactivation (100 °C for 10 minutes) and the protease trypsin as described before [16]. To rule out effects of contaminating DNA in HA samples, HA was treated with DNAse (10 Units of DNAse I for one hour at 37 °C followed by inactivation at 65 °C for 10 minutes, to remove trace amounts of DNA). To confirm LMW-HA induction of IL-8, HA samples were treated with 0.05 Units/2 µg sample HAdase (Streptomyces HAdase, Sigma Chemical Co., St. Louis, MO) at 60 ºC for 72 hours followed by inactivation at 65 °C for 10 minutes to remove HA.
Inhibitors of mitogen-activated protein kinase (MAPK) and NF-κB/IκB pathways
To determine the role of the MAPK and NF-κB/IκB pathways in LMW-HA induced IL-8 production, we used the extracellular signal-regulated kinase 1/2 (ERK 1/2) inhibitor U0126 (25 µM) the JNK II inhibitor SP600125 (10 µM), the NF-κB peptide inhibitor SN 50 and the NF-κB control peptide SN 50 inactive control (EMD Biosciences, La Jolla, CA). Cells were pre-incubated with inhibitors for one hour in serum-free media, followed by stimulation with HA for 6 hours. An LDH-based toxicology assay (Sigma Chemical Co, St. Louis, MO) was performed to rule out toxic effects of the inhibitors. No toxicity was found.
Immunoblot analysis
Cell lysates were normalised for protein concentration (BioRad, Inc., Hercules, CA) and resolved on a 10% bis-polyacrylamide gel, then transferred toImmunobilon-P membranes (Millipore Corp., Bedford, MA). Blots were incubated with antibody to p-JNK, p-ERK, p-p38, NF-κB/IκB, JNK, ERK and p38 (Santa Cruz Biotechnology, Santa Cruz, CA) (1:200) for two hours at room temperature and developed by enhanced chemiluminescence (NEN Life Science Products, Boston, MA). For NF-κB/IκB, the nuclear protein was separated from the cytosolic proteins as previously described [14].
Analysis of IL-8 protein
Cell culture supernatants were removed and centrifuged at 1200 g for 15 minutes to remove cellular debris. An enzyme-linked immunosorbent assay (ELISA) was used to measure IL-8 protein levels (R&D, Minneapolis, MN).
Statistical analysis
Statistical analyses were performed using Statview 4.5 (Abacus Concepts, Inc., Berkeley, CA). The concentration of IL-8 in cell supernatants was compared by analysis of variance (ANOVA) and then subsequent multiple comparisons were performed by the Scheffe-test. All values were expressed as mean ± standard error of the mean. Significance was determined by p <0.05.
Results
LMW-HA increased IL-8 production in BEAS-2B cells independent of contaminating LPS, protein or DNA
To address the hypothesis that LMW-HA production is pro-inflammatory, LMW-HA from the stretched IMR-90s was used, as previously described [1]. We previously showed that un-stretched IMR-90s only produce HMW HA [1]. In the current study, LMW-HA from bovine vitreous humor (fig. 1A) and LMW-HA isolated from stretched IMR-90 fibroblasts (fig. 1B), but not HMW-HA from rooster comb, significantly increased IL-8 protein in BEAS-2B cells (fig. 1A). HAdase treatment of HA preparations significantly blocked IL-8 production, whereas treatment of HA preparations with heat inactivation or protease to remove contaminating protein, or DNAse to remove contaminating DNA did not affect IL-8 production.
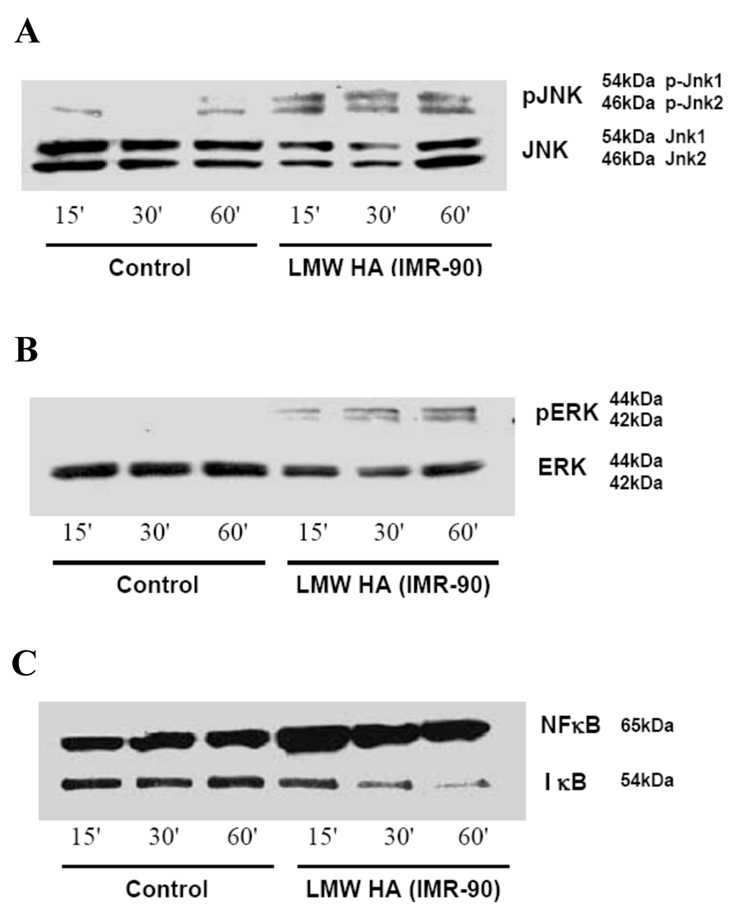
Figure 3
LMW-HA from stretched foetal lung fibroblasts (IMR 90 cells) activated the JNK, ERK, and NF-κB/IκB pathways in human BEAS-2B bronchial epithelial cells.
Western blot analysis: A. c-jun N-terminal kinase; B.ERK-1 and ERK-2; and C.NFκB in nuclear fraction/ IκB in cytosolic fraction. Each blot is representative of 3 separate experiments.
LMW-HA activated JNK, ERK and NF-κB/IκB pathways, but not the p38 pathway
Both forms of LMW-HA, from bovine vitreous (fig. 2A) and from stretched IMR-90s (fig. 3A), but not HMW HA from rooster comb (fig. 2A), increased phosphorylation of JNK (JNK 1 and JNK 2) as early as 15 minutes after exposure. This effect was sustained for up to 60 minutes with no change in the total JNK (JNK 1 and JNK 2) (fig. 2A and 3A). Both forms of LMW-HA (fig. 2B and 3B), but not HMW-HA (fig. 2B), increased phosphorylation of ERK 1 and ERK 2 with no change in total ERK. Both forms of LMW-HA (fig. 2C and 3C), but not HMW-HA (fig. 2C), also increased NF-κB expression in the nuclear fraction and decreased IκB expression in the cytosol, suggesting NF-κB activation. To determine the specificity of action of LMW-HA in the activation of MAPK kinase and NFκB/IκB pathways, HAdase treated HMW-HA from rooster comb and LMW-HA from bovine vitreous were included as controls. HAdase treated LMW-HA and HMW-HA did not increase JNK or NF-κB activation (fig. 2D). LMW-HA and HMW-HA did not activate the p38 pathway (data not shown).
IL-8 production by LMW-HA depended on the JNK and NF-κB/IκB pathways and, to a lesser extent, on the ERK pathway
Treatment with the JNK inhibitor SP600125, a reversible ATP-competitive inhibitor with greater than 20-fold selectivity relative to other kinases in inhibiting the action of p-JNK, but not the expression of p-JNK, significantly inhibited bovine vitreous LMW-HA-induced IL-8 production (fig. 4A). Inhibition of NF-κB activity with NF-κB inhibitor SN50, which inhibits translocation of the NF-κB active complex into the nucleus [17], significantly blocked the LMW-HA-induced IL-8 production compared to the NF-κB control peptide (fig. 4A). We observed only a partial inhibition of IL-8 production with ERK inhibition (fig. 4A). HMW-HA from rooster comb did not induce IL-8 production and this was not affected by inhibition of JNK or NF-κB activity (fig. 4A). Similar results were obtained regardless of whether the LMW-HA was derived from bovine vitreous humor (fig. 4A) or was collected from stretched IMR-90 fibroblasts (fig. 4B).
Discussion
Ventilator-associated lung injury (VALI) is acute lung injury that develops while a critically ill patient is mechanically ventilated [18]. Alveolar over-distention and cyclic atelectasis are known as the principal triggers of alveolar injury during positive pressure ventilation [19–21]. Therefore, all current ventilatory and pharmacologic strategies aim to reduce the incidence of VALI by preventing alveolar damage. However, even with these strategies, the mortality associated with VALI remains unacceptably high [22]. This suggests that other segments in the airway besides the alveoli might be affected by high-tidal volume ventilation and could contribute to VALI. Indeed, new evidence showed that high tidal volume mechanical ventilation – one of the culprits in VALI – results in substantial bronchial airway mechanical strain [13]. Our own observations had suggested a similar phenomenon [1]. The choice of transformed bronchial epithelial cells in the current study reflects our intent to understand the molecular and signalling underpinnings of the effects of high tidal volume mechanical ventilation in bronchial epithelium and its possible role in VALI.
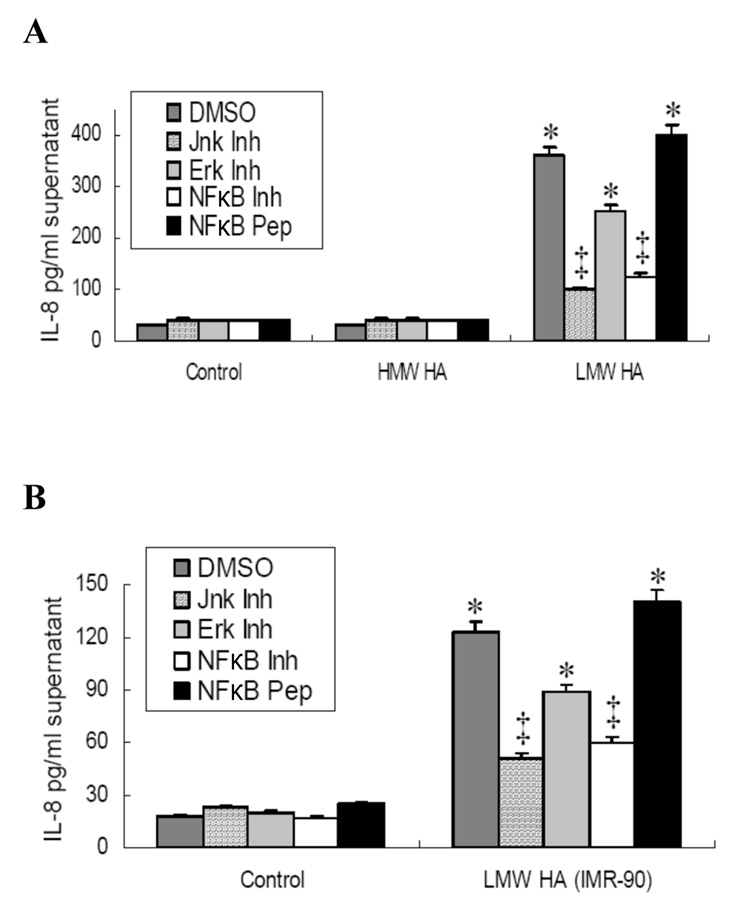
Figure 4
Inhibition of JNK and NF-κB, but not ERK, blocked LMW-HA induced IL-8 production in human BEAS-2B bronchial epithelial.
BEAS-2B cells were pre-incubated with inhibitors of JNK, ERK, and NF-κB or with an NF-κB control peptide for one hour before HA stimulation. BEAS-2B cells were then exposed to LMW-HA, HMW HA, or no HA with carrier DMSO in serum free medium for 6 hours.
A. LMW-HA from bovine vitreous humor and HMW HA from rooster comb (n = 5);
B. LMW-HA isolated from IMR-90 fibroblast HA (n = 3).
* p<0.05 vs controls; HMW HA; LMW-HA+JNK inhibitor and LMW-HA+NF-κB inhibitor.
‡ p<0.05 vs. LMW-HA+DMSO; LMW-HA+ERK inhibitor and LMW+NF-κB control peptide.
Our lab has been interested in studying glycosamoniglycans as pharmacologic targets in lung disease [23–25]. To this end, we have studied the pro-inflammatory properties of HA, a common lung extracellular matrix component [26]. HA is synthesised by HA synthases (HAS) [27–29] and it exists in a HMW form (>1000 kDa). However, it is the low molecular weight form (LMW-HA) that is pro-inflammatory and recent evidence revealed that LMW-HA is a pro-inflammatory molecule in the lung: LMW-HA (200 kDa) isolated from the serum of patients with acute lung injury, stimulated MIP-2 production in macrophages through binding to Toll-like receptors (TLR) 2 and 4 [12]. In addition, the over-stretched lung produces LMW-HA that up-regulates IL-8 production in a HAS 3 dependent manner [6]. In some animal models of lung injury, the production of HA starts a host innate immunity response that leads to the production of more inflammatory molecules and the mobilisation of cellular components that are critical for the resolution of the injury. This however, could lead to more tissue damage.
The size of HA that induces cytokine production appears to vary and may depend on the structure of the HA produced. It was found that LMW-HA of 180 kDa and 370 kDa was produced in a rat model of VALI [6]. The measurement of the size of HA can be difficult and the measured size represents an average size, since the fragments of HA in a sample are not uniform. Despite any variation that may have occurred in our measurement of the size of the HA isolated from the stretched lung fibroblasts, there was a clear difference in LMW-HA <500 kDa and HMW-HA >1000 kDa. Since LMW-HA can be generated by degradation of HMW-HA forms or by the direct synthesis of HAS 3, we cannot rule out that the breakdown of HMW-HA to LMW-HA may also occur in vivo. However, HAS 3 deficient mice did not produce LMW-HA when exposed to high tidal volume ventilation, indicating that at least in this form of acute lung injury the breakdown of HMW-HA to LMW-HA was not important [6].
The downstream signalling mechanism of LMW-HA involves the binding to HA receptors, called hyaladherins [30]. The hyaladherins include CD44, the receptor for HA-mediated motility (RHAMM), TLR2, TLR4, layilin, LYVE-1 and HARE [31]. CD44 is a trans-membrane glycoprotein and it is the major receptor for HA [32]. LMW-HA cross-links with CD44 and activates the signal transduction pathway that causes activation of transcription factors and chemokine production. In the current study, it was shown that LMW-HA synthesised by human lung fibroblasts, induced IL-8 production in human bronchial epithelial cells, via JNK, ERK1/2, and NF-κB/IκB pathways leading to AP-1 and NF-κB translocation to the nucleus.
We have previously shown – in vitro and in vivo – that JNK activation is critical in the pathogenesis of VALI and the current observations confirm the role played by the extracellular signal-regulated kinases in its pathogenesis [15, 33]. Therefore, the findings presented here suggest that two mechanisms of JNK and ERK activation might be in play during VALI. Firstly, JNK and ERK are activated due to their mechano-sensitive nature, and secondly, JNK and ERK could perpetuate lung damage when turned on by stretch-induced LMW-HA.
The synchronised activation of AP-1 and NF-κB by the same effecter is in line with previous observations. The concurrent mobilisation of both transcription factors was reported in human hepatoma cells in response to hepatitis B virus [34], in mouse dendritic cells in response to nickel sulphate [35], and in primary human lung fibroblasts in response to Tumour Necrosis Factor (TNF)-α stimulation [36]. Similarly, the induction of IL-8 production and other pro-inflammatory cytokines by the simultaneous nuclear translocation of AP-1 and NF-κB has been reported in the past. Yasumoto reported that TNF-α and interferon (IFN)-γ synergistically induced IL-8 production in a human gastric cell line through the activation of AP-1 and NF-κB [37].
In relation to HA, other investigative groups have reported activation of AP-1 and NF-κB by HA. Boodoo et al. showed that HA fragments induced production of the IL-8 and IP-10 in human mucoepidermoid carcinoma cells (NCI-H292) and was dependent on the activation AP-1 and NF-κB, respectively [9]. Our findings may differ from those of Boodoo et al. for very specific reasons. Firstly, the cell lines used to investigate AP-1 and NF-κB activation in both experiments were different (NCI-H292 v. BEAS-2B). Secondly, the size of the HA fragments might have affected the end-point. The current study reports HA fragments of <500 kDa in size (that we called LMH-HA), while the size of the HA fragments on the aforementioned study was never reported (although it is understood to be LMW-HA). Finally, the measurement of IP-10 was never an end-point in the current study. Despite these differences, both studies are in agreement with the premise that LMW-HA can induce a pro-inflammatory cascade in the lung through AP-1 and NF-κB activating pathways.
Conclusion
The current findings confirmed a role for LMW-HA in the biotrauma theory of VALI. Therefore, we conclude that LMW-HA produced in an in vitromodel of VALI induces IL-8 production via AP-1 and NF-κB transcriptional mechanisms. LMW-HA plays an important role in lung inflammation and could provide a new target for the prevention and treatment of VALI [4].
Acknowledgements:Thanks to Marcella M. Mascarenhas Ph.D and Bin OuyangM.D./Ph.D,from the First Affiliated Hospital of Sun Yat-Sen University, Guangzhou, China, and Kuan-Jen Bai M.D from the Division of Pulmonary and Critical Care Medicine, Department of Medicine, Wang-Fang Hospital, Taipei Medical University, Taipei, Taiwan, for their technical assistance.
References
1 Mascarenhas MM, Day RM, Ochoa CD, Choi WI, Yu L, Ouyang B, et al. Low molecular weight hyaluronan from stretched lung enhances interleukin-8 expression. Am J Respir Cell Mol Biol. 2004;30(1):51–60.
2 Liu YY, Lee CH, Dedaj R, Zhao H, Mrabat H, Sheidlin A, et al. High-molecular-weight hyaluronan – a possible new treatment for sepsis-induced lung injury: a preclinical study in mechanically ventilated rats. Crit Care. 2008;12(4):R102.
3 Laurent TC, Fraser JR, Laurent UB, Engstrom-Laurent A. Hyaluronan in inflammatory joint disease. Acta Orthop Scand Suppl. 1995;266:116–20.
4 Garg HG, Quinn DA, Mascarenhas MM, Hales CA. Hyaluronan in Ventilator-Induced Lung Injury. In Chemistry and Biology of Hyaluronan. Edited by Garg HG, Hales CA. Boston: Elsevier; 2004:271.
5 Bracke KR, Dentener MA, Papakonstantinou E, Vernooy JH, Demoor T, Pauwels NS, et al. Enhanced Deposition of Low Weight Hyaluronan in Lungs of Cigarette Smoke-Exposed Mice. Am J Respir Cell Mol Biol. 2009.
6 Bai KJ, Spicer AP, Mascarenhas MM, Yu L, Ochoa CD, Garg HG, et al. The role of hyaluronan synthase 3 in ventilator-induced lung injury. Am J Respir Crit Care Med. 2005;172(1):92–8.
7 Nettelbladt O, Hallgren R. Hyaluronan (hyaluronic acid) in bronchoalveolar lavage fluid during the development of bleomycin-induced alveolitis in the rat. Am Rev Respir Dis. 1989;140(4):1028–32.
8 Zaman A, Cui Z, Foley JP, Zhao H, Grimm PC, Delisser HM, et al. Expression and role of the hyaluronan receptor RHAMM in inflammation after bleomycin injury. Am J Respir Cell Mol Biol. 2005;33(5):447–54.
9 Boodoo S, Spannhake EW, Powell JD, Horton MR. Differential regulation of hyaluronan-induced IL-8 and IP-10 in airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2006;291(3):L479–86.
10 Horton MR, Burdick MD, Strieter RM, Bao C, Noble PW. Regulation of hyaluronan-induced chemokine gene expression by IL-10 and IFN-gamma in mouse macrophages. J Immunol. 1998;160(6):3023–30.
11 Horton MR, Shapiro S, Bao C, Lowenstein CJ, Noble PW. Induction and regulation of macrophage metalloelastase by hyaluronan fragments in mouse macrophages. J Immunol. 1999;162(7):4171–6.
12 Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, et al. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med. 2005;11(11):1173–9.
13 Sinclair SE, Molthen RC, Haworth ST, Dawson CA, Waters CM. Airway strain during mechanical ventilation in an intact animal model. Am J Respir Crit Care Med. 2007;176(8):786–94.
14 Schaffer JL, Rizen M, L’Italien GJ, Benbrahim A, Megerman J, Gerstenfeld LC, et al. Device for the application of a dynamic biaxially uniform and isotropic strain to a flexible cell culture membrane. J Orthop Res. 1994;12(5):709–19.
15 Li LF, Ouyang B, Choukroun G, Matyal R, Mascarenhas M, Jafari B, et al. Stretch-induced IL-8 depends on c-Jun NH2-terminal and nuclear factor-kappaB-inducing kinases. Am J Physiol Lung Cell Mol Physiol. 2003;285(2):L464–75.
16 Ochoa CD, Baker H, Hasak S, Matyal R, Salam A, Hales CA, et al. Cyclic stretch affects pulmonary endothelial cell control of pulmonary smooth muscle cell growth. Am J Respir Cell Mol Biol. 2008;39(1):105–12.
17 Ishibashi Y, Nishikawa A. Role of nuclear factor-kappa B in the regulation of intercellular adhesion molecule 1 after infection of human bronchial epithelial cells by Bordetella pertussis. Microb Pathog. 2003;35(4):169–77.
18 Oeckler RA, Hubmayr RD. Ventilator-associated lung injury: a search for better therapeutic targets. Eur Respir J. 2007;30(6):1216–26.
19 Parker JC, Townsley MI, Rippe B, Taylor AE, Thigpen J. Increased microvascular permeability in dog lungs due to high peak airway pressures. J Appl Physiol. 1984;57(6):1809–16.
20 Gattinoni L, Pesenti A, Avalli L, Rossi F, Bombino M. Pressure-volume curve of total respiratory system in acute respiratory failure. Computed tomographic scan study. Am Rev Respir Dis. 1987;136(3):730–6.
21 Dreyfuss D, Soler P, Basset G, Saumon G. High inflation pressure pulmonary edema. Respective effects of high airway pressure, high tidal volume, and positive end-expiratory pressure. Am Rev Respir Dis. 1988;137(5):1159–64.
22 Petrucci N, Iacovelli W. Lung protective ventilation strategy for the acute respiratory distress syndrome. Cochrane Database Syst Rev 2007, (3)(3):CD003844.
23 Yu L, Quinn DA, Garg HG, Hales CA. Cyclin-dependent kinase inhibitor p27Kip1, but not p21WAF1/Cip1, is required for inhibition of hypoxia-induced pulmonary hypertension and remodeling by heparin in mice. Circ Res. 2005;97(9):937–45.
24 Thompson BT, Spence CR, Janssens SP, Joseph PM, Hales CA. Inhibition of hypoxic pulmonary hypertension by heparins of differing in vitro antiproliferative potency. Am J Respir Crit Care Med. 1994;149(6):1512–7.
25 Hassoun PM, Thompson BT, Steigman D, Hales CA. Effect of heparin and warfarin on chronic hypoxic pulmonary hypertension and vascular remodeling in the guinea pig. Am Rev Respir Dis. 1989;139(3):763–8.
26 Anderson Bray B. Hyaluronan in the Pulmonary Alveolus and Interstitium. In Chemistry and Biology of Hyaluronan. Edited by Garg GA, Hales CA. Boston: Elsevier; 2004:247.
27 Spicer AP, Augustine ML, McDonald JA. Molecular cloning and characterization of a putative mouse hyaluronan synthase. J Biol Chem. 1996;271(38):23400–6.
28 Shyjan AM, Heldin P, Butcher EC, Yoshino T, Briskin MJ. Functional cloning of the cDNA for a human hyaluronan synthase. J Biol Chem. 1996;271(38):23395–9.
29 Watanabe K, Yamaguchi Y. Molecular identification of a putative human hyaluronan synthase. J Biol Chem. 1996;271(38):22945–8.
30 Johnson P, Ruffell B. CD44 and its role in inflammation and inflammatory diseases. Inflamm Allergy Drug Targets. 2009;8(3):208–20.
31 Day RM, Mascarenhas M. Signal Transduction Associated with Hyaluronan. In Chemistry and Biology of Hyaluronan. Edited by Garg GA, Hales CA. Boston: Elsevier; 2004:153.
32 McDonald B, McAvoy EF, Lam F, Gill V, de la Motte C, Savani RC, et al. Interaction of CD44 and hyaluronan is the dominant mechanism for neutrophil sequestration in inflamed liver sinusoids. J Exp Med. 2008;205(4):915–27.
33 Li LF, Yu L, Quinn DA. Ventilation-induced neutrophil infiltration depends on c-Jun N-terminal kinase. Am J Respir Crit Care Med. 2004;169(4):518–24.
34 Kanda T, Yokosuka O, Nagao K, Saisho H. State of hepatitis C viral replication enhances activation of NF-κB- and AP-1-signaling induced by hepatitis B virus X. Cancer Lett. 2006;234(2):143–8.
35 Cruz MT, Goncalo M, Figueiredo A, Carvalho AP, Duarte CB, Lopes MC. Contact sensitizer nickel sulfate activates the transcription factors NF-κB and AP-1 and increases the expression of nitric oxide synthase in a skin dendritic cell line. Exp Dermatol. 2004;13(1):18–26.
36 Seidel P, Merfort I, Tamm M, Roth M. Inhibition of NF-kappaB and AP-1 by dimethylfumarate correlates with down-regulated IL-6 secretion and proliferation in human lung fibroblasts. Swiss Med Wkly. 2010;140:w13132.
37 Yasumoto K, Okamoto S, Mukaida N, Murakami S, Mai M, Matsushima K. Tumor necrosis factor alpha and interferon gamma synergistically induce interleukin 8 production in a human gastric cancer cell line through acting concurrently on AP-1 and NF-κB-like binding sites of the interleukin 8 gene. J Biol Chem. 1992;267(31):22506–11.