DOI: https://doi.org/10.4414/smw.2011.13270
Plasma sodium imbalance is the most common electrolyte disorder in clinical medicine [1–3]. The prevalence of mild hyponatraemia was reported to be as high as 15–30% in hospitalised patients. Hypernatraemia is much less frequent and occurs in 1–3% of all inpatients. Hypernatraemia reflects a net water loss or a hypertonic sodium gain and is relatively easy to diagnose. Since it is, however, associated with a high morbidity and mortality [4], it is crucial that adequate treatment is rapidly initiated. Hyponatraemia, particularly if of rapid onset, is also associated with substantial morbidity and mortality [1, 5, 6]. Hyponatraemia is classified as hypervolaemic, hypovolaemic or euvolaemic, based on the volume status of the patient [7]. The diagnostic work up to differentiate between varying aetiologies of hyponatraemia is based on complex clinical algorithms including ambiguous clinical signs. Thus, the diagnosis of hyponatraemia based on clinical signs and routine laboratory evaluation has a limited sensitivity and specificity of <50% [8, 9]. Accordingly, as few as 10% of physicians reach the correct diagnosis of the underlying cause of hyponatraemia with routinely used diagnostic algorithms [10]. Inadequate therapy as a result of misdiagnosis may have significant clinical consequences [9, 11]. Therefore, a readily available, accurate marker for the differential diagnosis of serum sodium imbalance and fluid status would be of major clinical importance. The most important osmotic and volume regulated hormone is arginine vasopressin (AVP). The amount of plasma AVP is regulated by osmotic and non-osmotic stimuli [12]. In hyponatraemia, AVP levels are either appropriately or inappropriately elevated in relation to osmolar status. In hypernatraemia, AVP is high except in the case of central diabetes insipidus. The measurement of AVP is cumbersome. It therefore remains unclear whether there is a cut off differentiating appropriate and inappropriate AVP increases and whether AVP measurement is helpful in the differential diagnosis of dysnatraemia. AVP derives from a larger precursor peptide (pre-provasopressin) which consists of a signal peptide, AVP, Neurophysin II and copeptin [13]. Copeptin is produced in equimolar amounts to AVP [14] and recent data show that copeptin levels mirror AVP levels [15, 16]. It has been demonstrated that copeptin shows identical changes during disordered water states as previously shown for AVP [16]. As compared to AVP, copeptin is very stable in plasma and serum and can be easily determined [14, 17, 18]. Recently, it has been shown that plasma copeptin is a suitable surrogate of AVP secretion in hyponatraemic disorders and could be helpful in the differential diagnosis of isolated hyponatraemia in otherwise healthy patients [19]. Different biomarkers were shown in clinical practice to improve the ability to diagnose, risk-stratify and manage patients [20]. Therefore, the current study aims to compare copeptin levels in serum sodium imbalance of different aetiologies in two important cohorts of inpatients with acute medical diseases admitted to the emergency department.
For the purpose of this study, we took advantage of data of three prospective studies performed between December 2002 and November 2007 at the University Hospital Basel. Patients with lower respiratory tract infections (study A), community-acquired pneumonia (study B) and acute cerebrovascular events, i.e. acute haemorrhagic stroke, acute ischaemic stroke and transient ischaemic attack (study C) on admission were included. In all patients, copeptin levels, sodium and osmolality levels were measured and fluid status was assessed.
The study design of these two randomised studies has been described in detail elsewhere [21, 22]. In brief, these studies included patients with suspected lower respiratory tract infection (study A) [21] and community-acquired pneumonia (study B) [22], respectively, hospitalised from December 2002 until April 2003 and from November 2003 until February 2005 in our hospital. Patients had to be admitted with a suspected lower respiratory tract infections [21] or community-acquired pneumonia [22], respectively. In both studies a total of 545 Patients were included and analysed. The primary endpoint was the use and length of treatment with antibiotic drugs using procalcitonin guidance as compared to standard recommended guidelines in lower respiratory tract infections and community-acquired pneumonia [21, 22]. A predefined secondary endpoint was the prognostic value of biomarkers such as copeptin to assess the severity and to predict outcome in patients with lower respiratory tract infections and community-acquired pneumonia [23]. The Pneumonia Severity Index (PSI) was calculated to assess severity of pneumonia in patients with community-acquired pneumonia [24]. Baseline assessment included clinical data and vital signs, co-morbid conditions and routine blood tests on admission of the patients.
Between November 2006 and November 2007, all patients (n = 605) presenting at the emergency department of our hospital with a suspected cerebrovascular event (i.e. acute haemorrhagic stroke, acute ischaemic stroke or transient ischaemic attack) were evaluated. The study design of this prospective cohort study has been described in detail elsewhere (ClinicalTrials. gov number, NCT00390962) (25). A total of 509 patients was included, with a final diagnosis of an ischaemic (n = 362) or haemorrhagic stroke (n = 40) or transient ischaemic attack (n = 107). The primary endpoint of this study was the prognostic value of copeptin in predicting functional outcome and death and as well the value of copeptin as a diagnostic marker in disturbances of water homeostasis. To determine the severity of disability in the group of acute ischaemic stroke patients, the National Institute of Health Stroke Scale Score (NIHSS) was used [26]. Baseline assessment included medical history, clinical and laboratory items and standardised work up of stroke aetiology.
This study aimed to evaluate copeptin in disturbances of serum sodium imbalance in acutely ill and hospitalised patients.
In hypervolaemic hyponatraemia (i.e. congestive heart failure (CHF), cirrhosis or nephrosis) the stimulation of AVP secretion is due to a secondary haemodynamic stimulus and shows a moderate increase of AVP. Hypovolaemic hyponatraemia can be the result of diminished extracellular volume caused by diarrhoea, diuresis or third-space sequestration. In hypovolaemic states, decreased intravascular volume leads to impaired water excretion in response to decreased distal glomerular filtration delivery and by stimulates AVP secretion. In contrast, patients with SIADH, AVP levels are inappropriately elevated in relation to osmolar status [12, 27, 28].
In hypernatraemic patients with central diabetes insipidus plasma AVP levels are inappropriately low with respect to plasma osmolality. Nephrogenic diabetes insipidus is due to renal resistance to the antidiuretic effects of AVP with clearly increased AVP levels. In other causes of hypernatraemia (i.e. dermal or gastrointestinal losses, renal disease or iatrogenic hypertonic sodium gain) AVP plasma levels are markedly increased [12, 27].
Our hypothesis was that copeptin as a direct mirror of AVP might be helpful in differentiating the various aetiologies of hypo- and hypernatraemia. Therefore, we hypothesised that slight but significant differences in copeptin levels between the differential diagnoses of dysnatraemia might be found.
The determination of the underlying cause of hypo- or hypernatraemia was accomplished by a standardised structured diagnostic approach as follows:
In all patients the cause of hypo- or hypernatraemia was determined retrospectively using chart review by three independent board-certified endocrinologists.
Patients with hyponatraemia were classified according to plasma osmolality (measured directly or calculated) into hypo-, normo and hyperosmolar hyponatraemia. Patients with hypoosmolar hyponatraemia were further categorised according to extracellular fluid volume status as hypovolaemic, euvolaemic or hypervolaemic. In study C the fluid volume status was clinically assessed prospectively and in study A/B it was evaluated retrospectively using the data on physical examination on admission and different laboratory parameters such as haemoglobin concentration and haematocrit.
Furthermore, the differential diagnosis of hyponatraemia was assessed using a standardised clinical algorithm based on the detailed medical history including pharmacotherapy and special diet as well as different clinical and laboratory parameters [29, 30]. Patients were classified as having a syndrome of inappropriate antidiuretic hormone secretion (SIADH) if they fulfilled the diagnosis criteria described by Bartter & Schwartz [31].
Hypernatraemic patients were classified by a standardised clinical algorithm into hypotonic fluid loss, pure water loss and hypertonic sodium gain, respectively [32].
All laboratory parameters were sampled at the same time point at admission. Blood samples were obtained from an indwelling venous catheter. Results of the routine blood analyses were recorded. Copeptin levels were measured with a new chemiluminescence sandwich immunoassay with a lower detection limit of the assay of 0.4 pmol/L and a functional assay sensitivity of <1 pmol/L [14]. In healthy volunteers median copeptin plasma concentrations were 4.2 [IQR 1.0–13.8] pmol/L [18]. The serum osmolality was measured directly using freezing-point osmometry [33]. The freezing-point osmometry [33] was also used for determination of urine osmolality. In all three cohorts serum osmolality was calculated by including the main osmotic constituents in plasma using the formula: 2 x (sodium [mmol/L] + potassium [mmol/L]) + urea [mmol/L] + glucose [mmol/L].
According to the literature the definition of normal plasma sodium concentrations may vary from 138 to 142 mmol/L or 135 to 145 mmol/L depending on different clinical laboratories [6, 29, 32, 34]. For the purpose of this study, we used our in-house reference levels to define sodium dysbalance, i.e. 130 to 125 mmol/L for mild hyponatraemia, <125 mmol/l for severe hyponatraemia, 143 to 145 mmol/L for mild hypernatraemia and >145 mmol/L for severe hypernatraemia. However, all results remained similar if different reference levels were used.
To describe the populations we used descriptive statistics: discrete variables are expressed as frequency (percentage) and continuous variables as medians and interquartile ranges [IQR]. In a first step, we analysed the studies with respiratory tract infections and stroke separately because of differences in the study population, co-morbidities, endpoints and in the diagnostic work up among the cohorts. We used a hierarchical approach and first tested for any difference between subgroups with Kruskal-Wallis one-way ANOVA, and in case of a significant result, we used Dunn’s post-hoc test for multiple testing to identify the specific group differences. We report 95% confidence intervals (CIs) of the mean difference assuming a normal distribution of mean copeptin differences.
In a second step, we then combined the cohorts for an overall analysis using multivariable hierarchical regression adjusted for age and gender [35, 36]. This model accounts for within and between study variability by adding the study to the model as a random effect. Due to the skewed distribution, we log transformed copeptin levels before entering into the model.
All statistical tests were two-tailed, and P <0.05 was considered to indicate statistical significance. The statistical analysis was performed using Graph Pad Prism, Version 4.00 for Windows (Graph Pad Software, San Diego California, USA) and Stata version 9.2 (College Station, Texas).
Out of 545 patients included 58 (10.6%) had hyponatraemia (<131 mmol/L) and 20 patients (3.7%) hypernatraemia (>142 mmol/L). The prevalence of mild hyponatraemia (130–125 mmol/L) was 8.8% and the prevalence of more severe hyponatraemia (<125 mmol/L) was 1.8%. Hypernatraemia was less common with a prevalence of 2.6% for mild hypernatraemia (143–145 mmol/L) and 1.1% for severe hypernatraemia (>145 mmol/L).
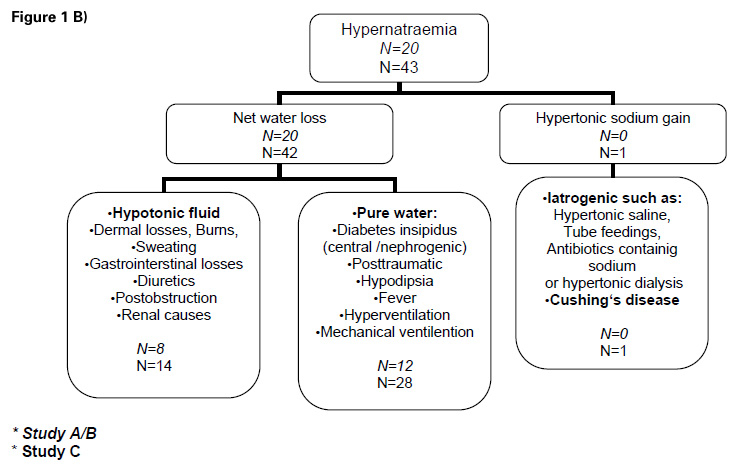
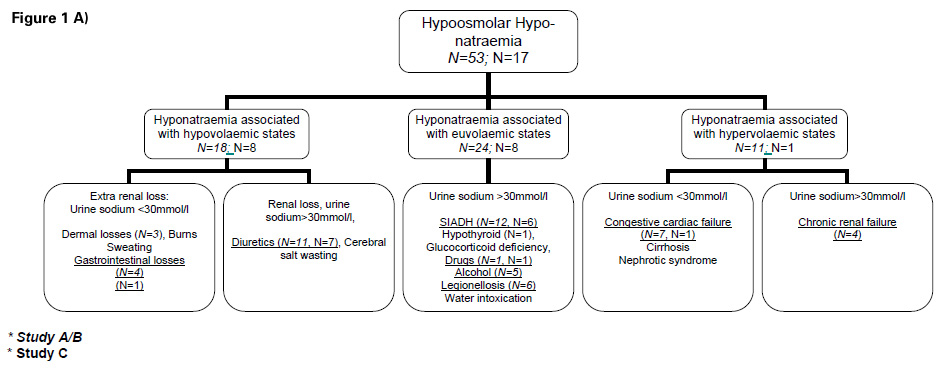
Figure 1
A) Flow sheet: Differential diagnosis hypoosmolar hyponatraemia
B) Flow sheet: Differential diagnosis hypernatraemia
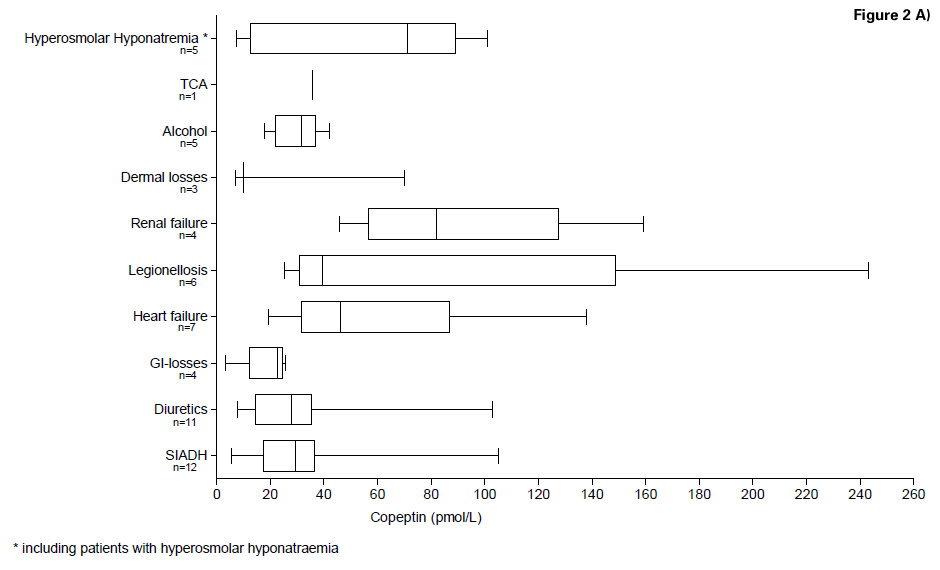
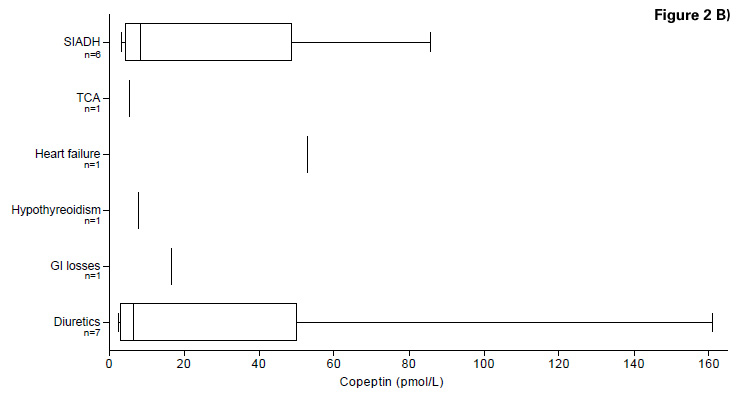
Figure 2
A) Copeptin levels in the differential diagnosis of hypoosmolar and hyperosmolar hyponatraemia (Study A/B)
B) Copeptin levels in the differential diagnosis of hypoosmolar hyponatraemia (Study C)
Classification of the patients according to diagnoses of hypoosmolar hyponatraemia is shown in figure 1A. Patients with hypernatraemia were classified according to aetiology as described in figure 1B. Baseline characteristics, including sex, age, clinical and laboratory parameters are shown in table 1.
In total 22 (4.3%) of the 509 patients had hyponatraemia and 43 (8.4%) hypernatraemia.
The aetiology of hypoosmolar hyponatraemia is described in figure 1A. Mild hyponatraemia (130–125 mmol/L) occurred in 3.5% of patients suffering an acute cerebrovascular event. More severe hyponatraemia (<125 mmol/L) was found in 0.8%. Hypernatraemic patients (n = 43) were classified in three different categories as shown in figure 1B. The prevalence of mild hypernatraemia (143–145 mmol/L) was 7.7% and that of severe hypernatraemia (>145 mmol/L) 0.8%. Detailed baseline characteristics of patients with hyponatraemia or hypernatraemia, respectively, including sex, age, clinical and laboratory parameters are shown in table 2.
Median copeptin level in all types of hyponatraemic patients with lower respiratory tract infections was 31.55 [IQR 22.0–50.4] pmol/L. We first tested the differences in copeptin levels across hypoosmolar patients with different volaemic states. Overall, there was a significant difference in median copeptin levels between patients with hypovolaemic hyponatraemia (n = 18, median 25.50 [IQR 9.02–29.65]), euvolaemic hyponatraemia (n = 24, median 31.90 [IQR 25.20–39.50]) and hypervolaemic hyponatraemia (n = 11, median 67.70 [IQR 45.30–96.20]) (Kruskal Wallis, p = 0.0012). After correction for multiple testing, this difference was only significant for patients with a hypovolaemic compared to hypervolaemic hyponatraemia (mean difference –45.19 (95%CI –71.78, –18.59), p <0.001) and for patients with hypervolaemic compared to euvolaemic hyponatraemia (mean difference –30.87 (95%CI –46.60, –3.84), p <0.05).
We then tested for differences in copeptin levels across different aetiologies of hyponatraemia. Again, there was evidence for significant differences (Kruskal Wallis, p = 0.02), but after adjustment for multiple testing, a significant difference in copeptin levels was only found between the group of patients with gastrointestinal losses (median 22.55 [IQR 12.31–24.70]) compared with the group of patients with renal failure (median 81.95 [IQR 56.70–127.60]) with a mean difference of –73.6 (95%CI –135.0, –12.3; p <0.05). Copeptin levels in the different groups are presented in table 1A and figure 2A.
Median copeptin level in hypernatraemic patients with lower respiratory tract infections was 48.0 [IQR 25.5–106.0] pmol/L. When testing for differences in copeptin levels across different causes of hypernatraemia, no significant difference was found (Kruskal Wallis, p = 0.70) (table 1B).
Median copeptin level in all types of hyponatraemic patients with acute cerebrovascular events (n = 22) was 8.5 [IQR 5.53–45.15] pmol/L. No significant difference was found in patients with hypovolaemic (n = 8), euvolaemic (n = 8) and hypervolaemic (n = 1) hyponatraemia (Kruskal Wallis, p = 0.466). In addition, no difference was found in the different aetiology groups (Kruskal Wallis, p = 0.59). Detailed results and copeptin levels are presented in table 2A and figure 2B.
Median copeptin level in patients with hypernatraemia and acute cerebrovascular events (n = 43) was 12.9 [IQR 4.60–40.20] pmol/L. Again, no significant difference was found in copeptin levels across different aetiologies of hypernatraemia (Kruskal Wallis, p = 0.26). Detailed results are presented in table 2B.
In a further step, we combined the different studies using hierarchical regression analysis adjusted for age and gender (table 3). For the hyponatraemia population, there was a significant association of hypervolaemic and copeptin levels (regression coefficient 0.17, 95%CI 0.04, 0.31) indicating that hypervolaemic patients have 0.17 pmol/L higher (log transformed) copeptin levels compared to patients without hypervolaemic. When focusing on the different aetiologies, heart failure (regression coefficient 0.42, 95%CI 0.09, 0.76) and renal failure (0.53, 95%CI 0.09, 0.97) were both associated with higher copeptin levels, compared to the other causes.
For the hypernatraemic population, no significant associations were found with regard to volaemia and different aetiologies (data not shown).
Copeptin levels increased with increasing severity of community-acquired pneumonia, classified according to the PSI score [24] (p <0.001). Median copeptin levels in patients with mild community-acquired pneumonia (defined as PSI class I to III) were significantly lower as compared to patients with severe community-acquired pneumonia (defined as PSI class IV and V) (21.0 [IQR 11.1–32.1] pmol/L vs. 44.5 [IQR 25.7–84.7] pmol/L, p <0.001).
Copeptin levels increased with increasing severity of stroke as defined by the NIHSS. Median copeptin concentration in patients with a NIHSS of 0-6 points (n = 217) was 8.6 (IQR 5.2–15.3) pmol/L, in patients with a NIHSS of 7–15 points (n = 90) 15.8 (IQR 7.7–28.7) pmol/L and in patients with a NIHSS >15 points (n = 55) 30.1 (IQR 9.0–67.9) pmol/L, p <0.0001).
| Table 1: Baseline characteristics study A/B: All Values are absolute numbers or medians and inter-quartile range A) Baseline characteristics of study A/B in patients with hyponatraemia | ||||||||||
| Hypoosmolar Hyponatraemia | Hyperosmolar Hyponatraemia | |||||||||
| Hypovolaemic | Euvolaemic | Hypervolaemic | ||||||||
| Diuretics | GI-losses | Dermal losses | SIADH | Alcohol | Legio- nellosis | TCA | Renal failure | Congestive heart failure | ||
| N=58 | 11 | 4 | 3 | 12 | 5 | 6 | 1 | 4 | 7 | 5 |
| Sex male/female (n) | 4/7 | 3/1 | 1/2 | 7/5 | 5/0 | 4/2 | 1/0 | 4/0 | 0/7 | 2/3 |
| Age-years | 83 (73-87) | 35 (26-40) | 76 | 63 (54-74) | 68 (60-82) | 41 (33-82) | 65 | 69 (57-80) | 86 (77-94) | 73 (62-83) |
| Laboratory parameters | ||||||||||
| Plasma sodium (mmol/L) | 127 (125-130) | 128 (127-129) | 129 | 129 (127-129) | 129 (125-130) | 129 (126-130) | 130 | 127 (126-129) | 127 (124-130) | 129 (124-130) |
| Plasma osmolarity mmol/Kg) | 279 (277-287) | 275 (273-276) | 275 | 276 (272-278) | 280 (274-283) | 278 (271-285) | 281 | 315 (294-328) | 277 (268-28) | 294 (287-313) |
| Creatinine (µmol/L) | 100 (82-116) | 110 (89-119) | 96 | 87 (74-137) | 85 (76-232) | 100 (82-168) | 49 | 480 (403-521) | 97 (76-151) | 105 (80-287) |
| Potassium (mmol/L) | 3.9 (3.4-4.3) | 3.7 (3.3-4.1) | 3.9 | 3.5 (3.0-4.0) | 3.9 (3.3-4.3) | 3.5 (2.8-3.9) | 4.3 | 4.4 (3.7-4.8) | 4.3 (3.9-4.8) | 4.5 (4.0-5.6) |
| Urea (mmol/L) | 8.3 (4.6-11.2) | 5.7 (3.7-6.3) | 5.5 | 5.6 (4.6-6.6) | 6.2 (4.7-9.4) | 6.6 (4.4-11.25) | 5.2 | 40.9 (24.9-56) | 7.3 (5.5-14.6) | 12.9 (6.6-23) |
| CRP (mg/L) | 143 (66-254) | 226 (108-321) | 110 | 232 (83-305) | 217 (158-239) | 251 (199-332) | 446 | 179 (110-216) | 202 (152-268) | 134 (60.5-164) |
| Leukocyte count (x109) | 11.4 (9.8-18.8) | 10.1 (7.2-16.7) | 15.0 | 10.8 (7.3-18) | 12.95 (8.75-25.6) | 11.1 (7.8-17.5) | 2.6 | 22.7 (13.5-32.9) | 16.9 (12.3-18) | 11.7 (8.5-14.02) |
| Copeptin (pmol/L) | 27.95 (14.62-35.15) | 22.55 (12.31-24.70) | 10.10 | 29.4 (17.4-36.5) | 31.7 (21.9-37) | 39.5 (31-148.8) | 35.6 | 82 (56.7-127.6) | 46.2 (31.4-86.9) | 71.2 (12.6-89.3) |
| Disease severity: | N | N | N | N | N | N | N | N | N | N |
| PSI 1-3 | – | 3 | 2 | 8 | 2 | 3 | – | 1 | – | – |
| PSI 4-5 | 9 | – | 1 | 4 | 3 | 3 | 1 | 3 | 6 | 5 |
| B) Baseline characteristics of study A/B in patients with hypernatraemia | ||
| Hypotonic fluid loss | Pure water loss | |
| N = 20 | 8 | 12 |
| Sex male/female (n) | 6/2 | 8/4 |
| Age-years | 79 (61-88) | 73 (64-82) |
| Laboratory parameters | ||
| Plasma sodium (mmol/L) | 144 (143-146) | 144 (143-147) |
| Plasmaosmolarity mmol/Kg) | 315 (308-321) | 312 (307-332) |
| Creatinine (µmol/L) | 123 (109-142) | 107 (76-165) |
| Potassium (mmol/L) | 4.3 (4.1-5.0) | 4.0 (3.7-4.2) |
| Urea (mmol/L) | 9.0 (6.0-13.6) | 10.4 (5.8-25.6) |
| CRP (mg/L) | 109 (2.4-187) | 113 (41-172) |
| Leukocyte count (x109) | 10.29±3.2 | 10.02 (7.6-12.35) |
| Copeptin (pmol/L) | 48.6 (24.4-168) | 49 (26.35-84.65) |
| Disease severity: | N | N |
| PSI 1-3 | 2 | 3 |
| PSI 4-5 | 5 | 9 |
| Table 2: Baseline characteristics study C: All Values are absolute numbers or medians and inter-quartile range A) Baseline characteristics of study C in patients with hyponatraemia | ||||||||
| Hypoosmolar Hyponatraemia | Normoosmolar Hyponatraemia | Hyperosmolar Hyponatraemia | ||||||
| Hypovolâemic | Euvolaemic | Hypervolaemic | ||||||
| Diuretics | GI-losses | SIADH | TCA | Hypo- thyroidism | Congestive heart failure | |||
| N = 22 | 7 | 1 | 6 | 1 | 1 | 1 | 2 | 3 |
| Sex male/female (n) | 1/6 | 0/1 | 0/6 | 0/1 | 0/1 | 1/0 | 0/2 | 3/0 |
| Age-years | 84 (70-90) | 69 | 76 (70-82) | 73 | 84 | 83 | 86 | 63 |
| Laboratory parameters | ||||||||
| Plasma sodium (mmol/L) | 129 (126-130) | 122 | 128 (127-130) | 119 | 121 | 128 | 128 | 130 |
| Plasmaosmolarity mmol/Kg) | 278 (271-281) | 259 | 277 (263-281) | 270 | 265 | 274 | 287 | 308 |
| Urine sodium (mmol/L) | 90 (58-137) | – | 84 (57-104) | 45 | 93 | 199 | 55 | 64 |
| Urine osmolarity (mmol/Kg) | 323 (226-453) | 378 | 444 (268-702) | 199 | 282 | 708 | 266 | 375 |
| Creatinine (µmol/L) | 68 (48-74) | 70 | 70 (44-83) | 148 | – | 72 | 97 | 67 |
| Potassium (mmol/L) | 3.6 (3.3-3.9) | 3.4 | 3.8 (3-4.2) | 4.8 | 3.7 | 4.5 | 4.1 | 4.0 |
| Urea (mmol/L) | 4.1 (3.6-5.2) | 3.9 | 3.9 (3.7-6.6) | 8.9 | 3.9 | 8.8 | 7.0 | 157.4 |
| CRP (mg/L) | 3 (3-30.5) | 3 (3-62.51 | 3 | 6 | 4 | 125 | 3.5 | |
| Leukocyte count (x109) | 7.46 (5.67-9.49) | 5.37 (2.84- 8.35) | 6.190 | 10.910 | 7.950 | 7.655 | 10.89 | |
| Copeptin (pmol/L) | 6.42 (3.12-50) | 16.7 | 8.5 (4.39-48.65) | 5.51 | 7.7 | 53 | 16.96 | 40.3 |
| Disease severity: | N | N | N | N | N | N | N | |
| Stroke: NIHSS 0-6 | 4 | 4 | 1 | – | – | – | – | |
| Stroke: NIHSS 7-15 | 1 | 1 | – | – | – | – | – | |
| Stroke: NIHSS >15 | – | 1 | – | – | 1 | – | 3 | |
| B) Baseline characteristics of study C in patients with hypernatraemia | |||
| Hypotonic fluid loss | Pure water loss | Hypertonic sodium gain | |
| N = 43 | 14 | 28 | 1 |
| Sex male/female (n) | 9/5 | 14/14 | 0/1 |
| Age-years | 76 (63-83) | 71 (59-82) | 71 |
| Laboratory parameters | |||
| Plasma sodium (mmol/L) | 144 (143-145) | 144 (143-145) | 143 |
| Plasmaosmolarity mmol/Kg) | 304 (299-310) | 306 (297-313) | 313 |
| Urine sodium (mmol/L) | 142 (82-162) | 111 (72-141) | – |
| Urine osmolarity (mmol/Kg) | 548 (415-642) | 518 (376-680) | – |
| Creatinine (µmol/L) | 70 (49-107) | 73 (65-86) | 62 |
| Potassium (mmol/L) | 3.8 (3.4-4.2) | 3.9 (3.7-4.2) | 3.7 |
| Urea (mmol/L) | 6.6 (4.9-8.7) | 5.8 (4.4-7.9) | 10.5 |
| CRP (mg/L) | 3 (3-5) | 3 (3-8) | 15 |
| Leukocyte count (x109) | 7.51 (5.82-8.66) | 7.76 (6.55-8.88) | 13.81 |
| Copeptin (pmolLˉ¹) | 22.6 (5.41-50.4) | 7.62 (3.85-33.3) | 18.00 |
| Disease severity: | N | N | N |
| Stroke: NIHSS 0-6 | 9 | 13 | 1 |
| Stroke: NIHSS 7-15 | 3 | 5 | – |
| Stroke NIHSS >15 | 1 | 5 | – |
| Table 3: Results from multivariate regression analysis combining the different studies A, B and C for the hyponatraemic population | ||
| A) Multivariate regression analysis combining the three studies for different volaemic states | ||
| Regression coefficient(95%CI) | p | |
| Hypovolaemia | –0.12 (–0.33, 0.08) | 0.228 |
| Hypervolaemic | 0.17 (0.04, 0.31) | 0.011 |
| B) Multivariate regression analysis combining the three studies for different aetiologies of hypoosmolar hyponatraemia | ||
| GI loss | –0.08 (–0.54, 0.37) | 0.723 |
| Acute heart failure | 0.42 (0.09, 0.76) | 0.013 |
| Hypothyroidism | –0.25 (–1.02, 0.51) | 0.517 |
| SIADH | 0.02 (–0.25, 0.29) | 0.872 |
| TCA | –0.10 (–0.66, 0.45) | 0.71 |
| Hyperglycaemia | 0.25 (–0.15, 0.64) | 0.219 |
| Legionellosis | 0.39 (–0.01, 0.79) | 0.053 |
| Alcohol induced | 0.07 (–0.34, 0.47) | 0.743 |
| Dermal losses | –0.10 (–0.58, 0.37) | 0.669 |
| Renal failure | 0.53 (0.09, 0.97) | 0.017 |
| Results from multivariable hierarchical regression analysis adjusted for age and gender and including the different studies as a random effect. Regression coefficients represent units of increase in log transformed copeptin levels. | ||
The principal finding of this study is that plasma levels of the AVP precursor peptide copeptin appear to add very little information to the work up of sodium imbalances in patients with acute diseases such as lower respiratory tract infections, community-acquired pneumonia or stroke admitted to the emergency department.
It has recently been demonstrated that copeptin is a new promising diagnostic tool in the differential diagnosis of otherwise healthy patients with isolated hypoosmolar hyponatraemia [19].
In our study, we included hospitalised patients with acute and stressful diseases such as lower respiratory tract infections, community-acquired pneumonia or acute cerebrovascular events. Thereby the copeptin levels of the patients with different aetiologies of hypoosmolar hyponatraemia were not statistically different. Yet, patients with hyponatraemia due to chronic renal failure compared to patients with gastrointestinal losses had higher copeptin levels. This result is in accordance with a recent published study of confounding factors with regard to interpretation of copeptin values. The authors showed that patients with impaired renal function had higher plasma levels of copeptin. This might be due on the one hand to the activation of the AVP system in patients with renal impairment and the high abundance of vasopressin receptors on renal tubules and on the other hand to the fact that copeptin is mainly cleared by the kidneys [37].
AVP has not only haemodynamic and osmoregulatory effects but also acts synergistically with corticotrophin releasing hormone activating the hypothalamo-pituitary-adrenal axis (HPA-axis) [38, 39]. Recently, we demonstrated that copeptin levels were significantly lower in non stressed healthy controls compared to hospitalised patients with mild and moderate levels or patients after large surgical interventions with high stress, thereby reflecting the individual stress level [39]. This suggests that even mild to moderate stress situations contribute to a remarkable copeptin release. Accordingly, previous studies showed increasing copeptin levels with increasing severity of disease in patients with community-acquired pneumonia [15, 23], stroke [25] and patients with cardiac insufficiency or myocardial infarction [40, 41]. In these studies, copeptin levels was an independent predictor of outcome [23, 25] reflecting the individual hypothalamic stress level and the degree of activation of the hypothalamo-pituitary-adrenal-axis. Accordingly, copeptin levels in our cohort of patients increased with increasing levels of disease severity.
Patients in study A/B had higher copeptin levels than patients in study C. A possible explanation is that the number of patients with a severe disease, defined with a PSI of IV to V was higher in study A/B compared to patients with a severe disease in study C (defined by a NIHSS (>15). Alternatively, the higher inflammatory stimulus in patients in study A/B compared to study C might have contributed to the higher copeptin levels. It has been shown that pro-inflammatory cytokines, such as interleukin-1-β (IL-1β), interleukin-6 (IL-6) and tumour necrosis factor-α (TNF-α), can activate the hypothalamic paraventricular cells to produce AVP [42].
The following limitations of our study must be taken into account. Firstly, investigating copeptin in the differential diagnosis of hypo- and hypernatraemia was not a predefined endpoint in study A/B. Thus, the differential diagnoses of hypo- and hypernatraemia were assigned retrospectively by chart review assessed by board certified endocrinologists. Secondly, the investigated study population encompassed only a small spectrum of acute diseases presenting at the emergency room of our hospital and therefore these diseases must be considered as an arbitrary selection. However, this selection of patients reflects frequent diseases known to be often accompanied by disturbances of sodium homeostasis [2, 3], making our findings clinically relevant. Thirdly, due to the limited number of patients with sodium imbalances and thus low statistical power, this study is at risk for both, type I errors (false positive) and type II errors (false negative results). Therefore our results should be interpreted with caution and validation in a larger cohort is paramount. Nevertheless our results should help to design new studies by taking into account the newly gained knowledge that probably the value of copeptin is restricted either to patients with no further acute illnesses beside their sodium imbalance or, in severely ill patients, possibly to patients with more severe hypo- or hypernatraemia. Fourthly, we did not measure AVP for a direct comparison with copeptin, mainly due to the analytical problems with AVP measurement [15, 18]. However, several studies have shown that copeptin is released in equimolar amounts to AVP directly mirroring AVP release [14–16]. In a study with 24 healthy volunteers, copeptin showed identical changes during disordered water states or osmolarity as previously shown for AVP [16].
In conclusion, measurement of plasma copeptin levels in patients with acute diseases appears to add very little information to the work up of sodium imbalances and levels are similar in varying causes of mild sodium imbalances. It is likely that the non-osmotic “stress”-stimulus in acute hospitalised patients is a major confounder and overrules the osmotic stimulus.
Acknowledgment: We thank the staff of the clinics of emergency medicine, internal medicine and endocrinology and the department of neurology as well as the department of clinical chemistry for most helpful support during the study.
1 Upadhyay A, Jaber BL, Madias NE. Incidence and prevalence of hyponatremia. Am J Med. 2006;119(7 Suppl 1):S30–5.
2 Rabinstein AA, Wijdicks EF. Hyponatremia in critically ill neurological patients. The neurologist. 2003;9(6):290–300.
3 Ellison DH, Berl T. Clinical practice. The syndrome of inappropriate antidiuresis. N Engl J Med. 2007;356(20):2064–72.
4 Borra SI, Beredo R, Kleinfeld M. Hypernatremia in the aging: causes, manifestations, and outcome. J Natl Med Assoc. 1995;87(3):220–4.
5 Smith DM, McKenna K, Thompson CJ. Hyponatraemia. Clin Endocrinol. 2000;52(6):667–78.
6 Reynolds RM, Seckl JR. Hyponatraemia for the clinical endocrinologist. Clin Endocrinol. 2005;63(4):366–74.
7 Patel GP, Balk RA. Recognition and treatment of hyponatremia in acutely ill hospitalized patients. Clin Ther. 2007;29(2):211–29.
8 Musch W, Thimpont J, Vandervelde D, Verhaeverbeke I, Berghmans T, Decaux G. Combined fractional excretion of sodium and urea better predicts response to saline in hyponatremia than do usual clinical and biochemical parameters. Am J Med. 1995;99(4):348–55.
9 Chung HM, Kluge R, Schrier RW, Anderson RJ. Clinical assessment of extracellular fluid volume in hyponatremia. Am J Med. 1987;83(5):905–8.
10 Hoorn EJ, Halperin ML, Zietse R. Diagnostic approach to a patient with hyponatraemia: traditional versus physiology-based options. Qjm. 2005;98(7):529–40.
11 Verbalis JG, Goldsmith SR, Greenberg A, Schrier RW, Sterns RH. Hyponatremia treatment guidelines 2007: expert panel recommendations. Am J Med. 2007;120(11 Suppl 1):S1–21.
12 Ball SGs Vasopressin and disorders of water balance: the physiology and pathophysiology of vasopressin. Ann Clin Biochem. 2007;44(Pt 5):417–31.
13 Morgenthaler NG, Muller B, Struck J, Bergmann A, Redl H, Christ-Crain M. Copeptin, a stable peptide of the arginine vasopressin precursor, is elevated in hemorrhagic and septic shock. Shock (Augusta, Ga. 2007;28(2):219–26.
14 Struck J, Morgenthaler NG, Bergmann A. Copeptin, a stable peptide derived from the vasopressin precursor, is elevated in serum of sepsis patients. Peptides. 2005;26(12):2500–4.
15 Jochberger S, Morgenthaler NG, Mayr VD, Luckner G, Wenzel V, Ulmer H, et al. Copeptin and arginine vasopressin concentrations in critically ill patients. J Clin Endocrinol Metab. 2006;91(11):4381–6.
16 Szinnai G, Morgenthaler NG, Berneis K, Struck J, Muller B, Keller U, et al. Changes in plasma copeptin, the c-terminal portion of arginine vasopressin during water deprivation and excess in healthy subjects. J Clin Endocrinol Metab. 2007;92(10):3973–8.
17 Morgenthaler NG, Struck J, Jochberger S, Dunser MW. Copeptin: clinical use of a new biomarker. Trends in endocrinology and metabolism: TEM. 2008;19(2):43–9.
18 Morgenthaler NG, Struck J, Alonso C, Bergmann A. Assay for the measurement of copeptin, a stable peptide derived from the precursor of vasopressin. Clin Chem. 2006;52(1):112–9.
19 Fenske W, Stork S, Blechschmidt A, Maier SG, Morgenthaler NG, Allolio B. Copeptin in the differential diagnosis of hyponatremia. J Clin Endocrinol Metab. 2008 Nov 4.
20 Mueller C, Muller B, Perruchoud AP. Biomarkers: past, present, and future. Swiss Med Wkly. 2008;138(15-16):225–9.
21 Christ-Crain M, Jaccard-Stolz D, Bingisser R, Gencay MM, Huber PR, Tamm M, et al. Effect of procalcitonin-guided treatment on antibiotic use and outcome in lower respiratory tract infections: cluster-randomised, single-blinded intervention trial. Lancet. 2004;363(9409):600–7.
22 Christ-Crain M, Stolz D, Bingisser R, Muller C, Miedinger D, Huber PR, et al. Procalcitonin Guidance of Antibiotic Therapy in Community-acquired Pneumonia: A Randomized Trial. Am J Respir Crit Care Med. 2006;174(1):84–93.
23 Muller B, Morgenthaler N, Stolz D, Schuetz P, Muller C, Bingisser R, et al. Circulating levels of copeptin, a novel biomarker, in lower respiratory tract infections. Eur J Clin Invest. 2007;37(2):145–52.
24 Fine MJ, Auble TE, Yealy DM, Hanusa BH, Weissfeld LA, Singer DE, et al. A prediction rule to identify low-risk patients with community-acquired pneumonia. N Engl J Med. 1997;336(4):243–50.
25 Katan M, Fluri F, Morgenthaler NG, Schuetz P, Zweifel C, Bingisser R, et al. Copeptin: a novel, independent prognostic marker in patients with ischemic stroke. Ann Neurol. 2009;66(6):799–808.
26 Brott T, Adams HP Jr, Olinger CP, Marler JR, Barsan WG, Biller J, et al. Measurements of acute cerebral infarction: a clinical examination scale. Stroke; a journal of cerebral circulation. 1989;20(7):864–70.
27 Verbalis JG. Disorders of body water homeostasis. Best practice & research. 2003;17(4):471–503.
28 Miller M. Hyponatremia and arginine vasopressin dysregulation: mechanisms, clinical consequences, and management. J Am Geriatr Soc. 2006;54(2):345–53.
29 Kumar S, Berl T. Sodium. Lancet. 1998;352(9123):220–8.
30 Reynolds RM, Padfield PL, Seckl JR. Disorders of sodium balance. BMJ (Clinical research ed. 2006;332(7543):702–5.
31 Schwartz WB, Bennett W, Curelop S, Bartter FC. A syndrome of renal sodium loss and hyponatremia probably resulting from inappropriate secretion of antidiuretic hormone. 1957. J Am Soc Nephrol. 2001;12(12):2860–70.
32 Adrogue HJ, Madias NE. Hypernatremia. N Engl J Med. 2000;342(20):1493–9.
33 Sweeney TE, Beuchat CA. Limitations of methods of osmometry: measuring the osmolality of biological fluids. Am J Physiol. 1993;264(3 Pt 2):R469–80.
34 Adrogue HJ, Madias NE. Hyponatremia. N Engl J Med. 2000;342(21):1581–9.
35 Thompson SG, Turner RM, Warn DE. Multilevel models for meta-analysis, and their application to absolute risk differences. Stat Methods Med Res. 2001;10(6):375–92.
36 Turner RM, Omar RZ, Yang M, Goldstein H, Thompson SG. A multilevel model framework for meta-analysis of clinical trials with binary outcomes. Stat Med. 2000;19(24):3417–32.
37 Bhandari SS, Loke I, Davies JE, Squire IB, Struck J, Ng LL. Gender and renal function influence plasma levels of copeptin in healthy individuals. Clin Sci (Lond). 2009;116(3):257–63.
38 Lightman SL. The neuroendocrinology of stress: a never ending story. J Neuroendocrinol. 2008;20(6):880–4.
39 Katan M, Morgenthaler N, Widmer I, Puder JJ, Konig C, Muller B, et al. Copeptin, a stable peptide derived from the vasopressin precursor, correlates with the individual stress level. Neuro endocrinology letters. 2008;29(3):341–6.
40 Kelly D, Squire IB, Khan SQ, Quinn P, Struck J, Morgenthaler NG, et al. C-terminal provasopressin (copeptin) is associated with left ventricular dysfunction, remodeling, and clinical heart failure in survivors of myocardial infarction. J Cardiac Fail. 2008;14(9):739–45.
41 Khan SQ, Dhillon OS, O’Brien RJ, Struck J, Quinn PA, Morgenthaler NG, et al. C-terminal provasopressin (copeptin) as a novel and prognostic marker in acute myocardial infarction: Leicester Acute Myocardial Infarction Peptide (LAMP) study. Circulation. 2007;115(16):2103–10.
42 Chikanza IC, Petrou P, Chrousos G. Perturbations of arginine vasopressin secretion during inflammatory stress. Pathophysiologic implications. Ann N Y Acad Sci. 2000;917:825–34.
Funding / potential competing interests: NGM is employed by B.R.A.H.M.S, the manufacturer of the copeptin-assay (B.R.A.H.M.S CT-proAVP LIA, B.R.A.H.M.S AG, Hennigsdorf/Berlin, Germany). BM has served as consultant and received payments from B.R.A.H.M.S to attend meetings, speaking engagements or research unrelated to this trial. MCC and MK received payments from B.R.A.H.M.S for speaking engagements. No funding was obtained from commercial sources for this study. The remaining authors have nothing to declare.
Authors’ contribution: *equally contributing last authors