Figure 1
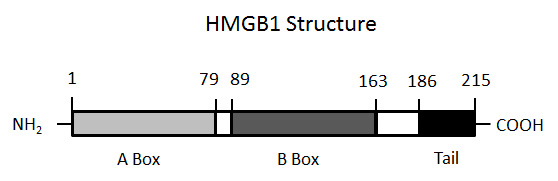
The structure of HMGB1. The figure illustrates the molecular structure of HMGB1, demonstrating the presence of two boxes (A and B) and the C-terminal tail.
DOI: https://doi.org/10.4414/smw.2011.13256
The role of HMGB1 and DAMP-PAMP complexes
Cell death is a ubiquitous process that marks the course of many diseases and occurs in diverse forms varying in mechanism, pattern and consequence. Of these diseases, immune-mediated diseases, including infectious, autoimmune and inflammatory syndromes, dramatically exemplify the role of cell death in pathogenesis, not just as an outcome but as an integral part of the process [1, 2].
In these conditions, immune system activation is invariably present, with products of dead cells in the blood and tissue serving as biomarkers as well as mediators in immunopathogenesis.
To elucidate its impact on the organism, cell death has long been dichotomised into two forms: apoptosis and necrosis. In this simplified schema, apoptosis corresponds to a programmed death process in which enzyme cascades lead to nucleolytic and proteolytic cleavage events along with cell shrinkage and fragmentation. In contrast, necrosis represents a sudden and disorderly form of death in which physical and chemical trauma irreversibly injures the cell, even causing its lysis. Whereas apoptosis can be physiological, necrosis is essentially always pathological [3–6].
While apoptosis and necrosis can be defined morphologically and biochemically, recent studies have added additional patterns to the repertoire of cellular extinction. The operation of these patterns may depend on the cell type, the inducing stimulus and downstream signalling pathways delivering the death blow to the cell [6]. In general, these patterns have been defined using in vitro systems; it is more difficult to identify patterns of cell death in vivo because of the advanced stages of pathological material as well as extensive clearance of dead cells by efferocytosis. Efferocytosis defines a process by which dead cells are removed by professional or non-professional phagocytes, of which macrophages are the most abundant and active [7, 8].
In general, apoptotic cells are rarely observed in clinical specimens although necrotic cells can be readily demonstrated in pathological specimens. Tumours, for example, are frequently filled with necrotic cells, making abnormal cell death in malignancy as much a feature of disease as abnormal cell growth [9, 10]. Among inflammatory diseases, leukocytoclastic vasculitis is defined by the presence of products of cell death (nuclear remnants or dust) from white cells that have undergone clasis (breaking apart). However, the pathway to that state of cellular destruction remains unknown, since biopsy can only reveal the end-stage of the process.
In the classification of cell death patterns, an important distinction concerns the impact of dead cells on the immune system: either positive or negative. Thus, studies have shown that cells dying by apoptosis are either devoid of immunological activity or may actually exert immunosuppressive properties. The paucity of immune reactivity of apoptotic cells may relate to the frequency with which apoptotic death occurs in physiological as well as in pathophysiological settings. Since apoptosis occurs during normal growth and development, its immunological “silence” may prevent the inadvertent generation of auto-reactivity to dead and dying cells which emerge by the billions each day [11, 12].
In contrast to the silent death of apoptosis, cell death by necrosis appears to be immunologically very noisy or, to use popular terms, dangerous or alarming. As shown in in vivoandin vitrosystems, cells undergoing necrosis can be potent immune stimulators. This activity likely results from the exposure or extracellular release of molecules termed DAMPs (damage associated molecular patterns) by analogy with PAMPs (pathogen associated molecular patterns). The structural diversity of DAMPs is striking, ranging from small molecules as simple as ATP and uric acid to large macromolecules such as heat shock protein, DNA or RNA [13].
The key feature of DAMPs is their ability to translocate from cells and stimulate both toll-like receptor (TLR) and non-TLR sensors in cells of the innate immune system. Reflecting this property, DAMPs also fit into the categories of danger molecules or alarmins. Alarmins are defined functionally by their ability to activate innate immunity, promote chemotaxis and stimulate antigen specific responses. While DAMPs have alarmin activity, certain pre-formed cytokines can display similar functional activity and fit into this terminology [14, 15].
Among other forms of cell death, NETosis can also generate inflammatory activity, especially in the setting of infection [16]. NETosis refers to a dramatic response of neutrophils to certain forms of immune stimulation. In NETosis, the entire nucleus of the cell is extruded in a high molecular weight form to generate neutrophil extracellular traps (NETs); NETs can ensnare bacteria and promote their killing by virtue of attached anti-microbiol molecules. The NET structure, reflecting its content of DNA, can also stimulate immune reactivity. This pattern of cell death appears to be specific for neutrophils although nuclear release can also occur with certain forms of necrosis.
Among recently defined processes, pyroptosis is a unique form of cell death which occurs in cells with metabolic and other forms of stress, and results from the activation of the inflammasome and generation of capase-1 activity. Since caspase-1 plays a key role in the maturation and secretion of IL-1β and IL-18, cells undergoing pyroptosis are a rich source of pro-inflammatory cytokines. IL-33, a molecule which has nuclear localisation, is also released in this form of death [17]. Interestingly, genetic abnormalities in the regulation of the inflammasome can give rise to auto-inflammatory syndromes, such as Muckle-Wells syndrome in which IL-1β plays a key role in inducing disease manifestations [18].
Finally, autophagy is a form of regulated death in which the cell digests itself to counteract stress and provide additional sources of energy to withstand this event. Although it can be a protective measure in settings of prolonged or intense stress such as starvation or infection, autophagy may be associated with cell death; in this case, cells may die withautophagy rather than from autophagy [19].
Given the array of cell death mechanisms and evidence that even necrosis can be regulated, investigators have questioned the role of this process in homeostasis. Among its consequences, cell death can deprive microorganisms (either bacterial or viral) of a home and certainly cell suicide can prevent replication of infecting organisms. In this conceptualisation, cell death can be an intrinsic feature of host defence, triggering inflammation and containing infection by removing local sanctuaries for intracellular multiplication. As such, the specific pathway of death may be less important than its efficiency in eliminating and combating infecting organisms.
An important determinant of the immune activity of any dying death cell is, in essence, its lifespan as an agonal entity. Compelling evidence indicates that cells dying by apoptosis undergo rapid clearance because of their display of molecules called “eat me” signals that interact with humoral and cellular systems to promote phagocytosis and removal by efferocytosis [7, 8, 20–22]. Hypotheses to explain this very rapid clearance are based on the idea that the persistence of dead cells can be dangerous since an apoptotic cell can transition to a late stage with pro-inflammatory properties; cells undergoing this transition are termed secondarily necrotic cells although their relationship to primary necrotic cells is not fully known [5].
Since apoptosis can occur in many physiological situations, rapid clearance may provide a fail-safe mechanism to prevent inflammation and auto-reactivity by removing cells which could become pro-inflammatory if they underwent a transition to secondary necrosis. As such, the life span of apoptotic cells in the body is very short. Indeed, abnormalities in the clearance process (e.g., complement deficiency) may be associated with the autoimmunity. In both humans and mice, for example, deficiency of C1q, a protein that can bind apoptotic cells, is highly associated with systemic lupus erythematosus (SLE) suggesting that impaired clearance of dead cells can stimulate both inflammation as well as drive autoantibody production [23, 24].
In contrast to apoptosis, necrosis is essentially always pathological and signalling of danger is necessary. Since the necrotic cell provides the basis for this signalling, the clearance process appears much more constrained than that which occurs with apoptotic cells. In this conceptualisation, slow clearance preserves a source of danger molecules to promote inflammation and signal repair.
As these considerations suggest, cells dying by necrosis represent an important element in host defence, signalling danger and inducing inflammation whether death results from infectious or non-infectious events. This signalling results in part from the release of intracellular molecules which, once freed from the usual confines of cells, can stimulate immune responses by interacting with both TLR and non-TLR sensors [25, 26]. The release process appears passive and the consequence of disrupted cell membrane integrity or actual cell lysis. Once the permeability barriers are down, the intracellular contents can translocate extracellularly, presumably by mass action down concentration gradients. Once in an extracellular location, intracellular molecules can act as DAMPs, driving inflammation in the local vicinity as well as stimulating the influx of inflammatory cells.
To understand endogenous stimulators of danger, investigators have characterised a variety of DAMPs or alarmins, focusing on their activity as isolated and purified entities on the assumption that they act as conventional cytokines or chemokines [13–15]. The results of these studies have been both interesting and puzzling since, in some instances, the activity of these molecules has been difficult to substantiate with highly purified products. Furthermore, the activity of these molecules has not been invariably correlated with their extracellular expression, suggesting that location is not sufficient to confer DAMP activity.
A further complexity in analysing the activity of DAMPs relates to the potential presence of lipopolysaccharide (LPS) or endotoxin in many systems, especially in in vivo animal models unless animals are kept germ-free. Since many molecules can bind LPS, it can be difficult to exclude the contribution of a PAMP to the activity of a DAMP. Even in in vitro systems, LPS contamination can complicate the interpretation of experiments since foetal calf sera and other media components can contain LPS. Ridding a system entirely of LPS is very difficult, making LPS contamination a constant worry especially if the DAMP is a charged molecule that can bind LPS.
The situation with HMGB1 clearly illustrates the difficulties in delineating DAMP activity, although, rather than confounding issues, the variability of HMGB1 activity and its interactions with PAMPs and cytokines may point to a fundamental mechanism by which this molecule works [27, 28]. Indeed, I would like to posit that just as many events inside immune cells during activation and death involve the assembly of complexes, so too, in the extracellular space, may the generation of multi-component complexes be essential to DAMP activity and its regulation. As such, molecules with DAMP activity may have structural features (e.g., charge, hydrophobic regions) that facilitate interaction with other molecules (proteins or nucleic acids) to create new moieties that can trigger responses.
Figure 1
The structure of HMGB1. The figure illustrates the molecular structure of HMGB1, demonstrating the presence of two boxes (A and B) and the C-terminal tail.
HMGB1 is a non-histone nuclear protein that is comprised of 215 amino acids that are arranged in two box structures (A box and B box) as well as a C terminal tail (fig. 1). HMGB1 can bind DNA, especially molecules with certain sequences or a bent structure, and can help organise chromosome architecture and regulate transcription. In the nucleus, HMGB1 is highly mobile since it is not tightly bound to chromatin; in some cells, HMGB1 can appear in the cytoplasm and shuttle back and forth between cellular compartments. Among factors governing its interactions and intracellular location, post-translational modifications of HMGB1 (i.e., acetylation) can modulate its properties [27–32].
The alarmin activity of HMGB1 was originally discovered in studies to identify novel mediators that are induced by LPS and therefore could contribute to sepsis. In seminal experiments, Wang et al. showed that the stimulation of macrophages with LPS leads to the abundant expression of HMGB1 in the media of cultures and, furthermore, that HMGB1 has immunostimulatory activity in vitro[33]. Subsequent studies established a role of HMGB1 in inflammatory responses in vivo. Thus, HMGB1 is elevated in settings of sepsis, shock and inflammatory arthritis, with strategies to reduce HMGB1 activity using antibodies or a competitive A box structure effective therapeutically [27, 28]. In in vivo settings such as sepsis, the expression of HMGB1 differs from that of a conventional cytokine such as TNF-α, appearing much later and having a sustained expression over time. Table 1 indicates diseases in which a role of HMGB1 has been proposed on the basis of animal models or studies indicating increased HMGB1 in either involved tissue or blood.
In the setting of immune stimulation, the extracellular expression of HMGB1 results from post-translational modifications including acetylation that influences its traffic from the nucleus to the cytoplasm [29–32]. Once in the cytoplasm, HMGB1 can enter endosomal vesicles for eventual secretion by a non-conventional mechanism. The release of HMGB1 is not confined to immune activation, however, since, when cells die, HMGB1 also translocates to the extracellular milieu [34, 35]. As such, HMGB1 can behave as a DAMP even though its extracellular release can occur in the setting of cell activation.
The release of HMGB1 during cell death is complicated since the extent of this process may depend on both cell type and the inducing stimulus. In original studies of the behaviour of HMGB1 during cell death, the release of this molecule, as assessed by Western blotting of the cell media and immunofluorescence microscopy, was demonstrated with necrotic cells but not with apoptotic cells. Furthermore, in cells undergoing apoptosis, the intracellular mobility of HMGB1 showed a dramatic change. As shown using FLIP (fluorescence loss in photobleaching) imaging, with apoptosis HMGB1 can become essentially immobile in the nucleus. Since treatment of apoptotic cells with trichostatin A (which blocks deacetylase activity) could allow HMGB1 release from apoptotic cells, these findings suggest that post-translational modifications (i.e., acetylation) can alter chromatin binding and nuclear retention [34].
The dichotomy in HMGB1 release during apoptosis and necrosis has a direct parallel with the immunological activity of apoptotic and necrotic cells, suggesting an important role of HMGB1 in determining the activity of dead cell phenotypes. Indeed, studies have indicated that HMGB1 expression could determine the immunological activity of dying cells. Thus, in in vitro culture systems, necrotic cells lacking HMGB1 (HMGB1-/-cells) fail to stimulate murine monocytes to produce TNF-α under conditions in which cells expressing HMGB1 are active. These findings suggest that the disposition of HMGB1 can determine the immune activity of dead and dying cells.
While this model had enormous appeal and reinforced the postulated dichotomy in immunological properties of death types, further experiments indicated a more nuanced situation. Thus, while original studies showed that apoptotic cells tightly retained HMGB1 in the nucleus, subsequent studies indicated that HMGB1 can translocate to the extracellular milieu as apoptosis proceeds [36–38]. While this stage may correspond to secondary necrosis, the release occurred readily. These finding raise questions about the extent and stability of an attachment of HMGB1 to chromatin during apoptosis. If HMGB1 is initially tightly bound in the nucleus during apoptosis, its subsequent release during secondary necrosis suggests a process of detachment as death proceeds or, alternatively, the presence of a more mobile pool, perhaps in the cytoplasm.
Another issue that arises in evaluating the dynamics of extracellular HMGB1 expression concerns the most appropriate models to elucidate the behaviour of macromolecules during death. Systems to study apoptosis are well-defined since this is a highly regulated process whose progress can be monitored by morphological and biochemical markers (e.g., caspase activation, DNA laddering). In contrast, necrosis results from physical or chemical trauma that causes cell destruction and lysis, although some forms of necrotic death (as well as non-apoptotic cell death forms described above) are more gradual and regulated.
In the absence of established and reliable models for necrosis, some investigators have used interventions such as freeze-thawing or even homogenisation to induce “necrosis.” While these interventions unquestionably kill cells rapidly, they can produce what is essentially a cell lysate. The amounts of protein (and other molecules) in such preparations are very high although the relationship between the amounts released during other forms of necrosis is not known. Nevertheless, the use of extreme measures of cell disruption to cause necrosis may lead to exaggerated measures of the amount of HMGB1 released during necrosis than would ordinarily occur.
While the relative amounts of HMBG1 released during apoptosis and necrosis remain a matter for future investigation, the immunological activity of HMGB1 in these death forms may differ nevertheless. Thus, HMGB1 released during apoptosis may have diminished immunological activity reflecting oxidation of key residues that can occur during redox disturbances in stressed cells [37]. In contrast, with necrosis, such residues may remain in their reduced form, maintaining activity. Among amino acids important in determining HMGB1, a cysteine at position 106 can influence interactions with TLR4 and create. Oxidation of this and other cysteines may affect the immunological activity of HMGB1 [39].
As these considerations suggest, the activity of HMGB1 may vary depending on whether it originates from activated, apoptotic or necrotic cells; these differences likely reflect the impact of post-translational modifications. The time course of the exposure of different forms of HMGB1 to the immune system may also vary, with trauma (depending on the inciting agent) potentially leading to more rapid HMGB1 release than immune activation.
Perhaps the most striking findings of the immune activity of HMGB1 concern the activity of highly purified preparations of HMGB1. As shown in a number of insightful studies, these preparations can be essentially devoid of activity although when mixed with either a TLR ligand such as LPS or a cytokine like IL-1, activity is restored [40–45]. Furthermore, complexing HMGB1 with DNA produces an immunologically active structure even though mammalian DNA itself lacks stimulatory properties. Together, these data suggest that HMGB1 itself is not a full-fledged alarmin but rather must act in partnership with another molecule. This molecule may or may not have immunological activity, with LPS and IL-1 partnering molecules whose activity can be boosted by association with HMGB1, while DNA partners molecules whose activity can be transformed by association. This array of interactions may account for the diversity of receptors implicated in HMGB1 activity. These receptors include TLR2, TLR4, TLR9 and RAGE (receptor for advanced glycation end-products) among others. Table 2 summarises the properties of HMGB1.
| Table 1: Diseases with a potential role of HMGB1 in their pathogenesis. |
| Sepsis |
| Haemorrhagic shock |
| Rheumatoid arthritis |
| Systemic lupus erythematosus |
| Polymyositis |
| Stroke |
| Traumatic brain injury |
| Table 2: Properties of HMGB1. |
| Non-histone nuclear protein |
| Mobile inside the cell nucleus |
| Binds DNA depending on sequence and structure |
| Redox sensitive |
| Interacts with cytokines, DAMPs and PAMPs |
| Stimulates innate immunity in the form of complexes |
As these considerations indicate, the immune properties of molecules like HMGB1 may be highly mutable and context-dependent, and as yet there is not an exact terminology to encompass their function. Indeed, the term dual function has been applied to molecules like HMGB1 that have both intracellular and extracellular activities. As now recognised, certain cytokines like IL-1α and IL-33 are also dual function molecules, binding to chromatin on the inside of the cell and stimulating immunity on the outside of the cell [46, 47]. HMGB1 is different from the nuclear cytokines, however, since it does not appear to have intrinsic immunological activity and requires another component to stimulate activity.
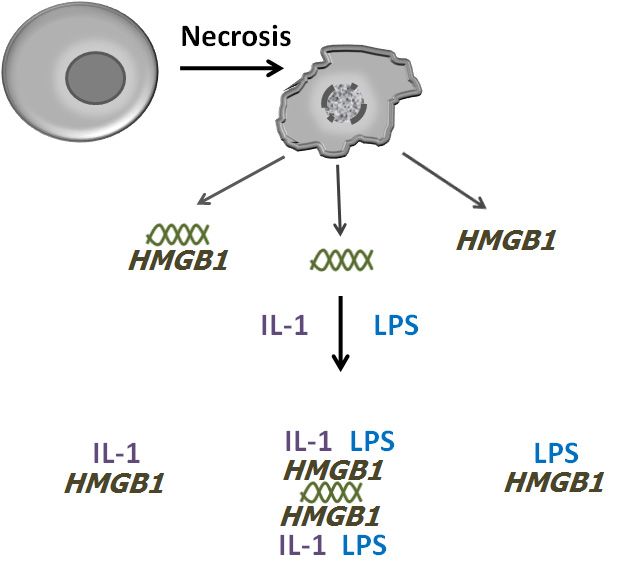
Figure 2
The formation of DAMP-PAMP complexes with HMGB1. The figure depicts a model by which HMGB1 can form complexes with DAMPs, PAMPs and cytokines following cell death. During necrosis in the setting of infection, both HMGB1 and DNA (illustrated by the double helix) can exit cells, alone or as a complex. Once in the extracellular milieu, HMGB1 can bind to molecules such as IL-1 and LPS and, depending on its interactions with DNA, can form complexes of varying structure and with a varying the number of components. The large complex illustrated is hypothetical although experimental data have demonstrated complexes of HMGB1 with DNA, IL-1 and LPS. This model allows the generation of many types of complexes that could vary depending on the components and the stoichiometry. For simplicity, the array of such complexes is not illustrated.
As a way of understanding HMGB1’s activity, I would like to posit that molecules like HMGB1 function as scaffolds or building blocks in the assembly of extracellular complexes which contain molecules that either have intrinsic immunology or can acquire such activity when in association with a binding molecule or as part of a large structure. This complex or nanostructure can be comprised of elements of both the blood proteome and the blood nucleome (the DNA and RNA in the circulation) that, in a concentration dependent fashion, aggregate to form structures that stimulate innate immunity [48]. In some instances, the components of the complexes may already be bound together on the inside of cells (e.g., HMGB1 and DNA) although these complexes may also form from these components on the outside of cells.
In this model, the binding of LPS is neither an artefact nor an experimental nuisance. Rather, it may be a key mechanism to accelerate events in innate immunity. During serious infection, time is of the essence to activate defensive measures. Thus, by binding circulating LPS or DNA, the immunological activity of the developing complex can rapidly increase, with the presence of the PAMP, even if in a low concentration, integral to the boost in immune potential. These complexes can be called DAMP-PAMP complexes. In addition to picking up circulating PAMPs to construct a DAMP-PAMP complex, HMGB1 can incorporate cytokines which, in initial stages of any response, are likely present in low concentration. With a complex decorated by PAMPs and cytokines, simultaneous interaction with a variety of receptor types can occur, acting synergistically to boost or amplify signals that would be much less powerful if delivered individually. Figure 2 illustrates this model.
These considerations do not imply that HMGB1 is the nidus for the formation of the DAMP complex but rather that it is an essential component which may even catalyse its formation because of its ability to bind diverse molecules. Whether HMGB1 undergoes any post-translational modifications extracellularly is not known but, if it is does, there would be a strong resemblance to the inflammasome or apoptosome which form as intracellular molecules come into contact with each other, frequently following phosphorylation events.
Over time, the size and composition of the complex can change. With effective host defence or antibiotic therapy, the levels of LPS may decline which in turn would lead to lower levels of cytokines. Similarly, as shock or ischemia abates the concentrations of HMGB1 would also fall and perhaps the complex or nanostructure would come apart. With the resolution of infection or the correction of metabolic imbalance, the system would return to the steady state with various components circulating in a monomeric form in insufficient amounts to interact or aggregate to generate the DAMP structure. While this model is speculative, it nevertheless provides a framework to understand the properties of molecules like HMGB1 and to explore their value as biomarkers as well as new targets of therapy.
1 Kono H, Rock KL. How dying cells alert the immune system to danger. Nature Rev Immunol. 2008;8:279–88.
2 Chen GY, Nuñez G. Sterile inflammation: sensing and reacting to damage. Nature Rev Immunol. 2010;10:826–37.
3 Silva MT, do Vale A, dos Santos NMN. Secondary necrosis in multicellular animals: an outcome of apoptosis with pathogenic implications. Apoptosis. 2008;13:463–82.
4 Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, et al. Classification of cell death: recommendations of the nomenclature committee on cell death 2009. Cell Death Differ. 2009;16:3–11.
5 Kalai M, Van Loo G, Vanden Berghe T, Meeus A, Burm W, Saelens X, et al. Tipping the balance between necrosis and apoptosis in human and murine cells treated with interferon and dsRNA. Cell Death Differ. 2002;9:981–4.
6 Antoine DJ, Williams DP, Kipar A, Laverty H, Park BK. Diet restriction inhibits apoptosis and HMGB1 oxidation and promotes inflammatory cell recruitment during acetaminophen hepatotoxicity. Mol Med. 2010;16:479–90.
7 Morimoto K, Janssen WJ, Fessler MB, McPhillips KA, Borges VM, Bowler RP, et al. Lovastatin enhances clearance of apoptotic cells (effercytosis) with implications for chronic obstructive pulmonary disease. J Immunol. 2006;176:7657–65.
8 Thorp E, Tabas I. Mechanisms and consequences of efferocytosis in advanced atherosclerosis. J Leukoc Biol. 2009;86:1089–95.
9 Gregory CD, Pound JD. Cell death in the neighbourhood: direct microenvironmental effects of apoptosis in normal and neoplastic tissues. J Pathol. 2011;223:177–94.
10 Voll RE, Hermann M, Roth EA, Stach C, Kalden JR, Girkontaite I. Immunosuppressive effects of apoptotic cells. Nature. 1997;390:350–1.
11 Sauter B, Albert ML, Francisco L, Larsson M, Somersan S, Bhardwaj N. Consequences of cell death: exposure to necrotic tumor cells, but not primary tissue cells or apoptotic cells, induces the maturation of immunostimulatory dendritic cells. J Exp Med. 2000;191:423–34.
12 Ferguson TA, Choi J, Green DR. Armed response: how dying cells influence T-cell functions. Immunol Rev. 2011;241:77–88.
13 Rubartelli A, Lotze MT. Inside, outside, upside down: damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol. 2007;28:429–36.
14 Harris HE, Raucci A. Alarmin(g) news about danger. Workshop on innate danger signals and HMGB1. EMBO. 2006;7:774–8.
15 Oppenheim JJ, Yang D. Alarmins: chemotactic activators of immune responses. Curr Opin Immunol. 2005;17:359–65.
16 Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176:231–41.
17 Bortoluci KR, Medzhitov R. Control of infection by pyroptosis and autophagy: role of TLR and NLR. Cell Mol Life Sci. 2010;67:1643–51.
18 Lachmann HJ, Quartier P, So A, Hawkins PN. The emerging role of interleukin-1β in autoinflammatory diseases. Arthritis Rheum. 2011;63:314–24.
19 Xu Y, Kim SO, Li Y, Han J. Autophagy contributes to caspase-independent macrophage cell death. J Biol Chem. 2006;281:19179–87.
20 Elliott MR, Ravichandran KS. Clearance of apoptotis cells: implications in health and disease. J Cell Biol. 2010;189:1059–70.
21 Poon IKH, Hulett MD, Parish CR. Molecular mechanisms of late apoptotic/necrotic cell clearance. Cell Death Differ. 2010;17:381–97.
22 Gregory CD, Pound JD. Microenvironmental influences of apoptosis in vivo and in vitro. Apoptosis. 2010;15:1029–49.
23 Taylor PR, Carugati A, Fadok VA, Cook HT, Andrews M, Carroll MC, et al. A hierarchical role for classical pathway complement proteins in the clearance of apoptotic cells in vivo. J Exp Med. 2000;192:359–66.
24 Muñoz LE, Janko C, Grossmayer GE, Frey B, Voll RE, Kern P, et al. Remnants of secondarily necrotic cells fuel inflammation in systemic lupus erythematosus. Arthritis Rheum. 2009;60:1733–42.
25 Zitvogel L, Kepp O, Kroemer G. Decoding cell death signals in inflammation and immunity. Cell. 2010;140:798–804.
26 Kono H, Karmarkar D, Iwakura Y, Rock KL. Identification of the cellular sensor that stimulates the inflammatory response to sterile cell death. J Immunol. 2010;184:4470–8.
27 Pisetsky DS, Erlandsson-Harris H, Andersson U. High-mobility group box protein 1 (HMGB1): an alarmin mediating the pathogenesis of rheumatic disease. Arthritis Res Ther. 2008;10:209.
28 Andersoon U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol. 2011;29:139–62.
29 Gardella S, Andrei C, Ferrera D, Lotti LV, Torrisi MR, Bianchi ME, et al. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO. 2002;3:995–1001.
30 Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A, et al. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO. 2003;22:5551–60.
31 Youn HJ, Shin J-S. Nucleocytoplasmic shuttling of HMGB1 is regulated by phosphorylation that redirects it toward secretion. J Immunol. 2006;177:7889–97.
32 Ito I, Fukazawa J, Yoshia M. Post-translational methylation of high mobility group box 1 (HMGB1) causes its cytoplasmic localization in neutrophils. J Biol Chem. 2007;282:16336–44.
33 Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–51.
34 Scaffidi P, Mistell T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–5.
35 Rovere-Querini P, Capobianco A, Scaffidi P, Valentinis B, Catalanotti F, Giazzon M, et al. HMGB1 is an endogenous immune adjuvant released by necrotic cells. EMBO. 2004;5:825–30.
36 Bell CW, Jiang W, Reich CF3rd, Pisetsky DS. The extracellular release of HMGB1 during apoptotic cell death. Am J Physiol Cell. 2006;291:C1318–25.
37 Kazama H, Ricci JE, Herdon JM, Hoppe G, Green DR, Ferguson TA. Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity. 2008;29:21–32.
38 Yamada Y, Fujii T, Ishijima R, Tachibana H, Yokoue N, Takasawa R, et al. The release of high mobility group box 1 in apoptosis is triggered by nucleosomal DNA fragmentation. Arch Biochem Biophys. 2011;506:188–93.
39 Yang H, Hreggvidsdottir HS, Palmblad K, Wang H, Ochani M, Li J, et al. A critical cysteine is required for HMGB1 binding to toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci USA. 2010;107:11942–7.
40 Li J, Wang H, Mason JM, Levine J, Yu M, Ulloa L, et al. Recombinant HMGB1 with cytokine-stimulating activity. J Intern Med. 2004;289:211–23.
41 Rouhianen A, Tumova S, Valmu L, Kalkkinen N, Rauvala H. Analysis of proinflammatory activity of highly purified eukaryotic recombinant HMGB1 (Amphoterin). J Leukoc Biol. 2007;81:1–10.
42 Sha Y, Zmijewski J, Xu Z, Abraham E. HMGB1 develops enhanced proinflammatory activity by binding to cytokines. J Immunol. 2008;180:2531–7.
43 Bianchi ME. HMGB1 loves company. J Leukoc Biol. 2009;86:573–6.
44 Hreggvidsdottir HS, Ostberg T, Wähämaa H, Schierbeck H, Aveberger AC, Klevenvall L, et al. The alarmin HMGB1 acts in synergy with endogenous and exogenous danger signals. J Leukoc Biol. 2009;86:655–62.
45 Urbonavicute V, Fürnrohr BG, Meister S, Munoz L, Heyder P, De Marchis F, et al. Induction of inflammatory and immune responses by HMGB1-nucleosome complexes: implications for the pathogenesis of SLE. J Exp Med. 2008;205:3007–18.
46 Moussion C, Ortega N, Girard JP. The IL-1 like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel ‘alarmin’? PLoS One. 2008;3:e3331.
47 Cohen I, Rider P, Carmi Y, Braiman A, Dotan S, White MR, et al. Differential release of chromatin-bound IL-1a discriminates between necrotic and apoptotic cell death by the ability to induce sterile inflammation. Proc Natl Acad Sci USA. 2010;107:2574–9.
48 Pisetsky DS, Ullal AJ. The blood nucleome in the pathogenesis of SLE. Autoimmun Rev. 2010;10:35–7.
Funding / potential competing interests: This work was supported by a VA Merit Review Grant and NIH Grants AI083923 and AI082402.