Diet and cancer
DOI: https://doi.org/10.4414/smw.2011.13250
Summary
Large claims have been made for the effectiveness of particular diets in preventing cancer or inhibiting its progression. However, more recent clinical studies have not confirmed this. Instead it seems that rather than specific dietary constituents, total calories influence cancer incidence and progression. In this review article, we summarise and interpret the available evidence for links between diet and cancer.
Introduction
Thirty years ago, Doll and Peto wrote an influential paper emphasising the contribution of lifestyle, diet in particular, on cancer incidence [1]. They estimated that one third of all cancer cases could be prevented by a healthier diet; a statement which was widely accepted in the scientific literature. Since then, a large number of studies and meta-studies have been published with varying and often contradicting results. Nonetheless, there is a general consensus that a “prudent” diet composed of mainly vegetables, fruit, whole grain and fish and a reduced intake of red meat, animal fat and refined sugar should be recommended, and that over-nutrition should be avoided.
In this review on the relationship between diet and cancer, we will, after a discussion of the molecular background and the methodological difficulties confronted by research in the field, present an overview about the effect of particular macro- and micronutrients on cancer incidence and consider the possible contribution of calorie reduction. We will conclude by presenting our view of what should and should not be recommended in order to reduce tumour incidence in the general population.
This review will be restricted to human studies, except when discussing calorie reduction. The physiological diets of laboratory animals, especially of mice and rats, are too different from human needs to justify the translation of results obtained in these studies into recommendations for people. Calorie reduction, on the other hand, seems to have comparable effects on all kinds of mammals.
Molecular mechanisms
For a long time, somatic mutagenesis stood at the centre of interest in the field of carcinogenesis. As a consequence, mutagenic dietary factors and nutrients that impair gene repair were investigated in detail (reviewed by [2, 3]). Among those identified are natural compounds consumed directly (e.g., in herbal teas) or indirectly via herbivores feeding on bracken fern for example. Food might also be contaminated with mutagenic molecules, with the best known example being aflatoxin produced by Aspergillus flavus growing on peanuts and cereals. Finally, heterocyclic amines, polycyclic aromatic hydrocarbons, N-nitroso-compounds or acrylamide can be formed by the process of cooking, particularly of meat [4]. The picture is complicated, however, by dietary factors that oppose the action of mutagens.
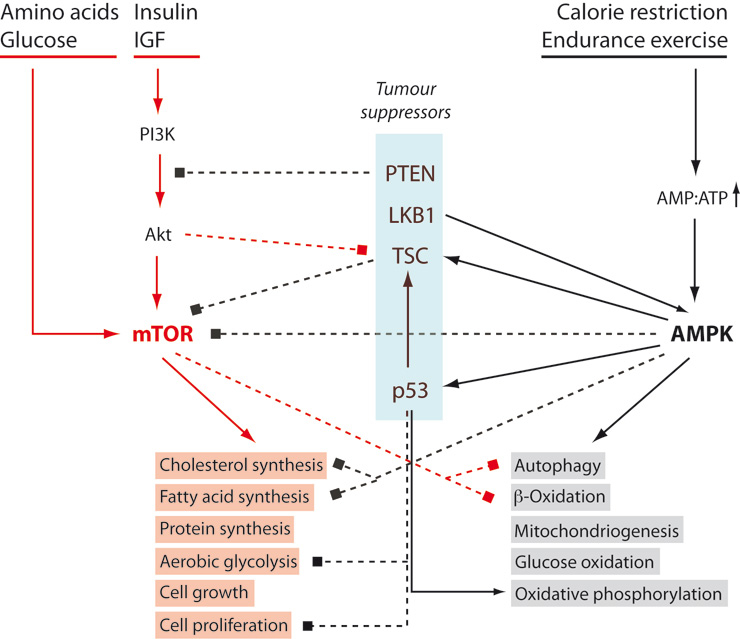
Figure 1
mTOR and AMPK oppose each other. Tumour enhancing pathways are coloured red, inhibitory interactions are represented by dashed lines. See text for details. AMPK: AMP-dependent kinase; IGF: insulin-like growth factor; LKB1: Serine/threonine-protein kinase 11; mTOR: mammalian target of rapamycin; PI3K: phosphoinositide 3-kinase; PTEN: phosphatase and tensin homologue; TSC: tuberous sclerosis complex.
More recently, the emphasis has shifted towards cancer-favouring and -inhibiting effects of the metabolic status of an individual. This shift of focus coincides with the rediscovery of the Warburg effect. In 1924, Warburg found that cancer cells mainly rely on aerobic glycolysis: they produce a large proportion of the adenosine triphosphate (ATP) they need by metabolising glucose to lactate which they secrete, even in the presence of sufficient oxygen [5]. Compared to normal cells, the aerobic, oxidative breakdown of pyruvate (from glucose) and fatty acids in mitochondria is reduced. Warburg originally thought that defective mitochondria were the cause of both aerobic glycolysis and carcinogenesis. However, while mitochondrial defects have been observed in some tumours, most harbour fully functional mitochondria.
In the last few years, our understanding of what affects the balance between the glycolytic phenotype distinctive of proliferating cells and the aerobic, oxidative phenotype of most normal cells has evolved considerably. At the same time, we are beginning to understand how an anabolic, glycolytic cellular mode might increase the probability of tumorigenesis. In the following, we will provide a broad overview that will serve as a framework for evaluating the possible effects of different diets.
In figure 1, we have placed two opposing hubs of the metabolic signalling network at the centre of events: the “mammalian Target of Rapamycin” (mTOR), a serine protein kinase which stimulates anabolic, ATP-consuming processes such as protein synthesis, lipid synthesis and cell growth (the “red block” in fig. 1); and the adenosine monophosphate (AMP)-dependent kinase (AMPK) which stimulates catabolic, ATP-generating processes, particularly in the mitochondrion (the “grey block” in fig. 1). Both are “energy sensors” with mTOR responding to high levels of energy carriers, glucose and amino acids, as well as to growth factors (insulin, insulin-like growth factor 1 etc.), and with AMPK responding to an increase of the AMP:ATP ratio (i.e. a slight depletion of the intracellular energy source ATP). The two pathways are not only activated by opposite metabolic situations, but they also inhibit each other.
It is clear that merely shifting the balance in favour of mTOR is not enough to cause cancer. However, it should be noted that four of the signalling intermediates are tumour suppressors that, directly or indirectly, inhibit mTOR: PTEN (phosphatase and tensin homologue), LKB1 (serine/threonine-protein kinase 11), TSC (tuberous sclerosis complex) and p53. It might, therefore, be expected that, in the long run, excessive mTOR activity will increase the probability of carcinogenesis.
High fasting blood glucose, insulin and insulin-like growth factor (IGF) concentrations stimulate the mTOR pathway and are also features of metabolic syndrome. The latter is known to positively correlate with cancer incidence, and we might expect that a diet preventing obesity and metabolic syndrome reduces the likelihood of developing cancer. But what appears to be straightforward in theory is often either hard to prove or not true. In the following, we will have a look at the evidence, after a discussion about the methodological difficulties of epidemiological studies on diet.
Although we have emphasised the role of mTOR pathways in carcinogenesis, we don't want to imply that this is the only mechanism explaining increased cancer incidence in obesity and the metabolic syndrome. There is, for example, abundant evidence that inflammatory adipokines released by visceral adipose tissue may favour tumourigenesis [6].
Methodological aspects
Establishing a causal relationship between a particular diet and cancer is difficult. Cancer is a chronic disease surfacing after many years of (possibly) harmful eating behaviour. This practically excludes the kind of controlled experiments demanded in science and leaves us with mainly observational studies.
Observational studies come in two flavours: In case-control studies, two groups are compared – one suffering from a disease (the “case”) and a matched control group. Eating habits are recorded with the help of questionnaires, but obviously suffer from recall bias, difficulties in remembering past behaviour and changes in that behaviour with time. Cohort studies are prospective. The diet of disease-free individuals is recorded, and their fate is followed over the years.
Both types of observational studies are flawed. Besides the fact that a correlation does at best suggest, but never prove a causal relationship, the groups are self-chosen and therefore most likely hide numerous additional differences in diet and lifestyle such as health-consciousness, physical activity and smoking. Those eating large amounts of red meat might represent the quintessential “steak and potato guys”. If a correlation is found, what causes it? Is it the red meat? Or the potatoes? Or maybe the combination of the two? Would a hypothetical group eating red meat and cabbage also be at risk? To a certain extent, these factors can be corrected for, but only when the observer is aware of their existence.
Often, case-control studies come up with a significant correlation between a given macro- or micronutrient and cancer incidence, but cohort studies fail to confirm the result. The reason for this is not clear, but the difficulty of recalling past eating behaviour may play a role [7, 8].
Not properly appreciated is the fact that many epidemiological studies which come up with a “significant” (p <0.05) relationship are wrong. In a paper provocatively entitled “Why Most Published Research Findings Are False”, Ioannidis explains why this is so. The likelihood of false positive results increases not only with small study sizes, small effects (many nutritional studies operate with relative risks of 1.1–1.5) and the prejudices of the investigators, but also with the number of possible correlational combinations typical for hypothesis-generating experiments [9].
Interventional studies (randomised controlled trials = RCTs) are more reliable because individuals are randomly assigned to treatment groups, excluding hidden biases introduced by self-choice (but see Martinez et al. [7] for a discussion of the pitfalls). However, participants have to stick to their assigned diet which limits the time-span of the trials.
What?
It is a very old and very common idea that by eating healthy food, cancer can be kept at bay. Many scientific and popular guidelines have been published that offer advice on what to eat, and worldwide, nutritional supplements worth 27 billion dollars are being sold every year. This stands in sharp contrast to the paucity of evidence, most of which is derived from epidemiological data. Controlled trials, on the other hand, are mostly missing. In this section, we discuss the evidence for pro- and anti-cancerogenic effects of different kinds of food.
Fat
A diet high in fat has been suspected to contribute to carcinogenesis and a poor outcome in cancer patients since the late 1950s. Lipids and lipid metabolites play a role in oncogenic signalling, and two preclinical examples are: (i) oxidised low density lipoprotein (LDL) promotes cellular transformation through NFκB signalling [10]; (ii) overexpression of monoacylglycerol lipase does increase migration, invasion and survival of cancer cells [11]. However, lipids and lipid metabolism in the body don’t directly reflect lipids in the diet – they are the result of a complex interplay between all dietary components, other lifestyle factors and genetics. Whether eating a high fat diet increases cancer incidence and mortality or not needs to be established by large, adequately powered, randomised and controlled interventional trials. No such study has yet been performed [12]. Nonetheless, case-control and cohort studies have shed some light on the question.
Colorectal cancer
Studies investigating the effect of total fat intake on colon cancer incidence have been inconsistent. Usually, case-control studies have been more likely to find a positive correlation than cohort studies. The Women’s Health Initiative randomised controlled trial found no such association. In most observational studies, the association between total fat intake and colorectal cancer incidence disappeared after an adjustment for total calories [12]. This may be due to the strong correlation between calorie and total fat intake in industrialised countries [13]. Interestingly, one cohort study even found a reduced incidence of colorectal cancer in women with a high consumption of high-fat dairy food [14]. The benefit might be mediated by conjugated linoleic acids, but there are many other compounds in milk whose influence on carcinogenesis is unaccounted for. In addition, there are some limited data that consumption of fish and w-3 long chain polyunsaturated fatty acids may be beneficial [12].
Prostate cancer
Several case-control and cohort studies have not been able to establish a link between total fat uptake and prostate cancer risk. There are not enough data to draw a conclusion regarding the effect of specific classes of fatty acids on prostate cancer incidence [15–20].
Breast cancer
The Women’s Health Initative study showed a risk reduction with a low fat diet, which was borderline significant [21]. However, other case-control and cohort studies have produced conflicting results [22, 23]. Again, it is total energy rather than total fat uptake that has an impact on the frequency of breast cancer [24].
Prompted by animal data, many case-control and cohort studies investigating the effect of specific fatty acids on breast cancer development have been performed. The results of these studies are prone to confounding effects due to the different possible sources of fatty acids [12]. Based on the current data, no firm conclusion can be drawn regarding the contribution of specific fatty acids to breast cancer incidence.
Taken together, in as much as fat uptake contributes to obesity, it may be correlated with a higher cancer risk. However, there is no convincing evidence that fat uptake per se (i.e. independent from total energy uptake and obesity) is a risk factor for malignant disease [12, 25].
Meat
Besides fat, meat (especially when red or processed) is counted among the "usual suspects" contributing to carcinogenesis [26]. However, current systematic reviews and meta-analyses do not support a causal role in colorectal or prostate cancer [27, 28]. In a recent meta-analysis of work on breast cancer, most case-control studies came up with a correlation between meat consumption and cancer, while the majority of cohort studies failed to confirm the link [29]. More recently, attention has shifted to meat fat as a potential detrimental factor. Freedman, for example, found a positive correlation with the development of hepatocellular carcinoma [30]. However, these data are also hampered by the correlation of fat and total energy. Randomised and controlled trials that could settle the issue are not available.
Fruits, vegetables and dietary fibre
Many case-control and cohort studies are dealing with the effect of fruits and vegetables on cancer incidence. Early data indicated a beneficial effect (summarised by Block et al. [31]) and, as recently as 2008, Freedman et al. found a reduced occurrence of head and neck cancers with increased fruit and vegetable consumption [32]. On the other hand, a Cochrane study came up empty when correlating the amount of dietary fibre with the occurrence of colorectal cancer [33]. Similarly, an Food and Drug Administration (FDA) review found little evidence to support an association between tomato consumption and cancer risk [34]. Lifestyle issues are powerful confounding factors when investigating the effect of fruits, vegetables and dietary fibre on health. Even after carefully adjusting for these factors, residual uncertainties remain [35].
Four large prospective trials have assessed the correlation between fruit and vegetable intake and overall cancer risk. In a European study, after 8.7 years of follow-up a higher intake of 200 g of vegetable and fruit resulted in a slightly reduced cancer risk (hazard ratio = 0.97, [36]). The result was significant, but not when subjected to a more stringent Bayesian analysis [37]. These results are in line with two earlier cohort trials that reported no correlation between fruit and vegetable uptake and cancer risk [38, 39]. In the NIH-AARP study, fruits or vegetables were not associated with cancer incidence, except for a small inverse association between vegetable consumption and cancer in men [40].
The inverse correlation between the uptake of fruits and vegetables and the consumption of alcohol and tobacco is a potent confounding factor. At this point, it has to be concluded that fruits, vegetables and dietary fibre have a very marginal, if any, effect on cancer incidence.
Vitamins, antioxidants and other micronutrients
Many trials have investigated the correlation between the consumption of vitamins and antioxidants and the incidence of cancer. In contrast to most other studies on diet and cancer, which rely heavily on a cohort or case-control design, some randomised controlled trials are available in the case of vitamins and antioxidants. In the Women’s Antioxidants Cardiovascular Trial, vitamin C, E and β-carotene were supplemented [41]. After a median follow up of 9.4 years, no difference in cancer incidence was noted. At the level of individual cancer types, a Cochrane review found no decrease in gastrointestinal cancers when vitamins and antioxidants were added to the diet [42]. In addition, multivitamins were not effective in preventing recurrence or increasing overall survival in colorectal cancer patients [43]. β-carotene did not prevent cancer, and on the contrary, it increased the risk of lung and gastric cancer in smokers and in asbestos workers (systematic review by Druesne-Pecollo [44]).
Based on a number of case-control and cohort studies, it was claimed that supplementing the diet with folic acid or natural folates may reduce the risk of colorectal cancer [45–47]. However, an effect was only seen in persons who took supplements for at least 15 years, whereas the number of polyps was not reduced. Heavy smokers and people who consume large quantities of alcohol, conditions known to deplete folate reserves, might benefit from folate supplements. In contrast, circulating folate was positively associated with the risk of prostate cancer in a meta-analysis of prospective studies showing an odds ratio of 1.18 (95% confidence interval 1.0–1.4 per 10 nmol/L increase) [48]. This suggests that there must be an ideal range of folate in the body, below and above which the cancer risk increases.
Selenium is another micronutrient studied for its cancer preventing potential. However, the SELECT trial has provided no evidence that selenium prevents cancer in the selenium replete population [49].
While in general, supplementation with vitamins, antioxidants or other micronutrients does neither reduce the risk of carcinogenesis nor improve the outcome in patients suffering from cancer, there is one possible exception: vitamin D was found to reduce the risk of colorectal cancer [50], reviewed in [51, 52]. In an unplanned subgroup analysis of the Women’s Health Initative, supplementation with calcium and vitamin D reduced the risk of melanoma in women with previous non-melanoma skin cancer [53]. However, with regard to breast cancer, prospective cohort trials showed a much smaller benefit than retrospective case-control studies [54]. In addition, most of the laboratory work which forms the rationale for studies on vitamin D was done with 1,25-dihydroxy-cholecalciferol, whereas in nutritional studies 25-hydroxy-cholecalciferol was supplemented. This complicates the interpretation of the results.
In summary, supplementation with vitamins, antioxidants or other micronutrients offers no relevant benefit in the primary prevention of cancer incidence and mortality. The potential benefits of vitamin D and folate have to be confirmed or disproved by additional studies, preferentially with interventional trials.
Alcohol
Although seldom fully appreciated, the relationship between alcohol consumption and cancer risk is firmly established [55]. Alcohol is the most frequent cause of hepatocellular carcinoma (HCC), accounting for about 40% of all cases. Regular consumption of more than 80 grams of alcohol per day for more than 10 years increases the risk for HCC approximately 5-fold, approaching an absolute risk of about 1% per year in alcoholic liver cirrhosis [56]. When adjusted for tobacco use and other potential confounding factors, alcohol consumption of more than 60 grams per day increases the relative risk for oral or hypopharyngeal squamous cell cancer 3.2 to 9.2 fold (reviewed by Goldstein et al. [57]). Drinking moderate quantities of alcohol (up to 12.5 g/day) results in a relative risk of 1.4 for esophageal cancer. The relative risk is 2.6 for intermediate-level (>12.5 to 50 g/day) and 5.5 for high-level (>50 g per day) alcohol consumption [58]. In colorectal cancer, there is a strong correlation with alcohol consumption as well. The relative risk is 1.21 (95% confidence interval 1.13–1.28) for moderate and 1.52 (95% confidence interval 1.27–1.81) for heavy (≥4 drinks per day) drinking [59]. Alcohol consumption also significantly increases the risk for breast cancer. The association is stronger for the hormone-sensitive form of breast cancer than for the insensitive form [60]. Consuming 3 or more alcoholic drinks per week also augments the risk of recurrence and cancer-related death upon diagnosis of breast cancer [61], but these results still await confirmation in other large, prospective studies of breast cancer survivors with long-term follow-ups. There also seems to be a modest increase in the incidence of prostate cancer if 7 or more drinks per day are consumed (reviewed by [62]).
Although the studies mentioned are case-control studies – for obvious reasons, controlled randomised trials are not feasable – the very high risk values and the consistency of the results allow us to conclude that confounding factors introduced by self-selection will not modify the interpretation substantially. Thus, ethanol and its metabolite acetaldehyde are carcinogens.
Tea and coffee
Green (unfermented) tea, Camellia sinensis, has a reputation for being healthy and reducing cancer risk. Limited preclinical and clinical data seem to support this belief [63]. In particular, a positive effect on the incidence of hepatocellular cancer was reported [64]. However, a recent Cochrane Review found no evidence for a cancer-prevention effect of green tea [65]. Interestingly, green tea consumption may contribute to weight loss or weight stabilisation [66]. Therefore, an indirect effect of green tea on cancer incidence, mediated by its supposed weight-lowering property, cannot be excluded.
Like green tea consumption, regularly drinking coffee has been associated with a reduced risk for hepatocellular cancer. These findings are supported by two meta-analyses of several recent case-control and cohort studies [67, 68]. Intriguingly, consumption of more than 3 cups of coffee per day was also associated with an improved virologic response to peginterferon and ribavirin in patients with hepatitis C [69]. This finding might provide a causal link between coffee and reduced hepatocellular cancer risk. However, there are no randomised controlled data on this issue. A meta-analysis of 13 cohort studies found no correlation between coffee consumption (more than 6 cups per day) and colorectal cancer risk [70]. The same analysis found a small positive correlation (relative risk = 1.28) between tea consumption (more than 4 cups per day) and colorectal cancer incidence. However, relatively few participants of the study consumed that much tea, and the type of tea consumed (black versus green) was not assessed. If there is a real effect of coffee or tea on cancer risk, it must be small and would be difficult to detect in epidemiological studies.
How much?
Obesity and other symptoms of metabolic syndrome are risk factors for cancer [6, 71–73]. Obesity increases the risk of colorectal cancer, particularly of the microsatellite-stable form [74]. In a nested, case-control study, obesity also correlated with a poor prognosis for prostate cancer patients, and preliminary data suggest that fatty acid synthase polymorphism might link obesity to mechanisms of tumour progression [75]. Obese breast cancer patients have a higher risk of tumour progression [76], and diabetes mellitus and hyperglycemia increase the hazard ratio for death in breast cancer patients [77, 78]. In summary, there is solid evidence that obesity has a limited but real influence on cancer incidence and mortality [79, 80].
As what, how often and how much an individual eats has a lot to do with whether he or she will become obese, dietary interventions that prevent people from becoming overweight will also reduce cancer incidence. However, rather than digress into the topic of how obesity may be prevented, we will discuss three dietary interventions that affect cancer incidence or progression favourably, at least in laboratory animals: calorie restriction, intermittent fasting and the ketogenic diet.
Calorie reduction
Calorie (or energy) restriction (CR) is defined as a reduction of calorie intake while maintaining sufficient levels of protein, essential fatty acids and micronutrients. CR is easily implemented in laboratory animals where baseline (ad libitum) intake is reduced by 20–40%. It should be noted that mice and rats kept round the clock with access to an unlimited food supply are overfed and “metabolically morbid” [81]. Energy-restricted animals resemble the wild state more closely.
Calorie restriction reduces the exposure of cells to high glucose levels, insulin and other growth factors, favouring a metabolic state dominated by AMPK, while at the same time inhibiting mTOR activity (fig. 1). It is no surprise, therefore, that calorie reduction does lower the frequency of spontaneous and induced tumours in laboratory animals (for references see [82–84]).
For obvious reasons, there is only circumstantial evidence that drastic calorie reduction has the same effect in humans. The example most often cited is a study of the population of Okinawa with a total energy intake 20% below the Japanese (and 40% below the US) average. They live longer, and death rates due to cancer are 30% lower than in the rest of Japan [85]. Another example is provided by individuals who experienced the “Dutch Hunger Winter” during World War II (1944–1945) and who had a lower risk of developing colorectal cancer later in life. In this case, unvoluntary severe CR, although of short duration, led to epigenetic changes that might explain the outcome [86].
It is impossible to impose substantial (20–40%) energy reduction regimes on human populations for a prolonged period of time. Healthy people subjected to this kind of experiment do not get used to feeling hungry, become apathic, cold-sensitive and sometimes depressive, as Benedict et al. described in a careful, book-length study published in 1919 [87]. It might even be dangerous, as discussed by Dirks et al. [88] and by Mattson who speculated, based on single observations and some animal experiments, that a causal link between a negative energy balance and the motor neuron disease Amyotrophic Lateral Sclerosis (ALS) might exist [89].
Diets calorie-reduced only by about 10% should be feasible and have been shown to improve certain risk factors [88]. However, no large long-term study on the effect of such a diet on cancer incidence has been published.
Intermittent fasting
Effects comparable to the ones observed when subjecting laboratory animals to calorie reduction have been obtained by “intermittent fasting”, that is increasing the intervals between meals. In the laboratory, feeding animals on alternate days (the “Every Other Day” diet or EOD) improves risk factors to the same extent as substantial calorie reduction, even though energy consumption is considerably higher [90]. Intermittent fasting also reduces chemically induced hepatocarcinogenesis [91] and delays spontaneous tumorigenesis in p53-deficient mice [92].
In the Western world, the three daily meals of the Seventies gradually gave way to today's herbivore-type pattern of almost continuous grazing [93]. In view of the results obtained with laboratory animals, careful prospective studies on the effect of reducing the number of meals on cancer incidence should be undertaken [94].
Ketogenic diet
Fasting (including intermittent fasting) induces ketogenesis, the production of the ketone bodies β-hydroxy-butyrate and acetoacetate from fatty acids in the liver. Another way of achieving high blood concentrations of ketone bodies is by eating a ketogenic diet: Diets low in carbohydrates and high in fat (>50% of the energy intake) are called ketogenic. They share a number of biochemical and biological effects with calorie reduction schemes [95], and it has been suggested that, like calorie reduction, they might have anti-tumour activity.
Ketogenic diets have been employed as adjuvant therapy in a few cases of patients with brain tumours. The reports were positive, but anectdotal [96, 97]. Although, at this time, it is not known whether the method is applicable to other kinds of tumours [98], we think that physicians should be aware of the results obtained so far and keep track of future developments.
Conclusions
If by changing our eating behaviour we can reduce the number of cancer patients, information on what is harmful and what might help will interest both individuals and society. However, we should beware of the cavalier attitude of “If it doesn’t help, at least it doesn’t do any harm” and not recommend diets with no proven benefit. Changing the eating behaviour of whole populations might have unforeseen effects when, for example, people replace something supposedly unhealthy with something equally bad or worse [99]. It may affect the economy, it may tempt legislators to introduce “incentives” or taxes that have no scientific basis, and last but not least, cancer patients might unnecessarily reproach themselves their past behaviour.
We have therefore subjected the often sanguine claims made in the past, mostly based on case-control studies, to a critical analysis comparing them to more recent and larger cohort studies and, if available, controlled randomised trials. These are our conclusions:
‒ Obesity leads to a limited but real increase of cancer risk and mortality, possibly due to an over-stimulation of mTOR-related pathways. Minimising those stimuli by limiting calorie-intake and the number of meals eaten per day is recommended.
‒ Alcohol is a carcinogen and increases cancer incidence and mortality.
‒ Consumption of fat or meat per se (independent of total calorie intake) probably does not increase the risk.
‒ Increasing the consumption of tea, coffee, fruits or vegetables is not expected to impact significantly on cancer rates, at least not in well-nourished populations.
‒ There is currently no compelling evidence that supplementing vitamins, antioxidants or other micronutrients reduces cancer incidence.
References
1 Doll R, Peto R. The causes of cancer: quantitative estimates of avoidable risks of cancer in the United States today. J Natl Cancer Inst. 1981;66:1191–308.
2 Ferguson LR. Role of dietary mutagens in cancer and atherosclerosis. Curr Opin Clin Nutr Metab Care. 2009;12:343–9.
3 Ferguson LR. Dietary influences on mutagenesis – where is this field going? Environ Mol Mutagen. 2010;51:909–18.
4 Zheng W, Lee S-A. Well-done meat intake, heterocyclic amine exposure, and cancer risk. Nutr Cancer. 2009;61:437–46.
5 Warburg O. über den Stoffwechsel der Carcinomzelle. Naturwissenschaften 1924;
6 Calle EE, Kaaks R. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer. 2004;4:579–91.
7 Martínez ME, Marshall JR, Giovannucci E. Diet and cancer prevention: the roles of observation and experimentation. Nat Rev Cancer. 2008;8:694–703.
8 Tsugane S, Sasazuki S. Diet and the risk of gastric cancer: review of epidemiological evidence. Gastric Cancer. 2007;10:75–83.
9 Ioannidis JPA. Why most published research findings are false. PLoS Med. 2005; 2: e124.
10 Hirsch HA, Iliopoulos D, Joshi A, et al. A transcriptional signature and common gene networks link cancer with lipid metabolism and diverse human diseases. Cancer Cell. 2010;17:348–61.
11 Nomura DK, Long JZ, Niessen S, Hoover HS, Ng S-W, Cravatt BF. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell. 2010;140:49–61.
12 Gerber M. Background review paper on total fat, fatty acid intake and cancers. Ann Nutr Metab. 2009;55:140–61.
13 Astorg P, Arnault N, Czernichow S, Noisette N, Galan P, Hercberg S. Dietary intakes and food sources of n-6 and n-3 PUFA in French adult men and women. Lipids. 2004;39:527–35.
14 Larsson SC, Bergkvist L, Wolk A. High-fat dairy food and conjugated linoleic acid intakes in relation to colorectal cancer incidence in the Swedish Mammography Cohort. Am J Clin Nutr. 2005;82:894–900.
15 Bidoli E, Talamini R, Bosetti C, et al. Macronutrients, fatty acids, cholesterol and prostate cancer risk. Ann Oncol. 2005;16:152–7.
16 Hedelin M, Chang ET, Wiklund F, et al. Association of frequent consumption of fatty fish with prostate cancer risk is modified by COX-2 polymorphism. Int J Cancer. 2007;120:398–405.
17 Liu X, Schumacher FR, Plummer SJ, Jorgenson E, Casey G, Witte JS. Trans-fatty acid intake and increased risk of advanced prostate cancer: modification by RNASEL R462Q variant. Carcinogenesis. 2007;28:1232–6.
18 Neuhouser ML, Barnett MJ, Kristal AR, et al. (n-6) PUFA increase and dairy foods decrease prostate cancer risk in heavy smokers. J Nutr. 2007;137:1821–7.
19 Park S-Y, Murphy SP, Wilkens LR, Henderson BE, Kolonel LN. Fat and meat intake and prostate cancer risk: The multiethnic cohort study. International Journal of Cancer. 2007;121:1339–45.
20 Wallstrom P, Bjartell A, Gullberg B, Olsson H, Wirfalt E. A prospective study on dietary fat and incidence of prostate cancer (Malmo, Sweden). Cancer Causes Control. 2007;18:1107–21.
21 Prentice RL, Caan B, Chlebowski RT, et al. Low-fat dietary pattern and risk of invasive breast cancer: the Women's Health Initiative Randomized Controlled Dietary Modification Trial. JAMA. 2006;295:629–42.
22 Thiebaut ACM, Kipnis V, Chang S-C, et al. Dietary fat and postmenopausal invasive breast cancer in the National Institutes of Health-AARP Diet and Health Study cohort. J Natl Cancer Inst. 2007;99:451–62.
23 Wang J, John EM, Horn-Ross PL, Ingles SA. Dietary fat, cooking fat, and breast cancer risk in a multiethnic population. Nutr Cancer. 2008;60:492–504.
24 Schulz M, Hoffmann K, Weikert C, Nothlings U, Schulze MB, Boeing H. Identification of a dietary pattern characterized by high-fat food choices associated with increased risk of breast cancer: the European Prospective Investigation into Cancer and Nutrition (EPIC)-Potsdam Study. Br J Nutr. 2008;100:942–6.
25 Leosdottir M, Nilsson PM, Nilsson J-A, Månsson H, Berglund G. Dietary fat intake and early mortality patterns – data from The Malmö Diet and Cancer Study. J Intern Med. 2005;258:153–65.
26 Ferguson LR. Meat and cancer. Meat Sci. 2010;84:308–13.
27 Alexander DD, Mink PJ, Cushing CA, Sceurman B. A review and meta-analysis of prospective studies of red and processed meat intake and prostate cancer. Nutr J. 2010;9:50.
28 Alexander DD, Miller AJ, Cushing CA, Lowe KA. Processed meat and colorectal cancer: a quantitative review of prospective epidemiologic studies. Eur J Cancer Prev. 2010;19:328–41.
29 Taylor VH, Misra M, Mukherjee SD. Is red meat intake a risk factor for breast cancer among premenopausal women? Breast Cancer Res Treat. 2009;117:1–8.
30 Freedman ND, Cross AJ, McGlynn KA, et al. Association of meat and fat intake with liver disease and hepatocellular carcinoma in the NIH-AARP cohort. J Natl Cancer Inst. 2010;102:1354–65.
31 Block G, Patterson B, Subar A. Fruit, vegetables, and cancer prevention: a review of the epidemiological evidence. Nutr Cancer. 1992;18:1–29.
32 Freedman ND, Park Y, Subar AF, et al. Fruit and vegetable intake and head and neck cancer risk in a large United States prospective cohort study. Int J Cancer. 2008;122:2330–6.
33 Asano T, McLeod RS. Dietary fibre for the prevention of colorectal adenomas and carcinomas. Cochrane Database Syst Rev 2002; CD003430.
34 Kavanaugh CJ, Trumbo PR, Ellwood KC. The U.S. Food and Drug Administration's evidence-based review for qualified health claims: tomatoes, lycopene, and cancer. J Natl Cancer Inst. 2007;99:1074–85.
35 Key TJ. Fruit and vegetables and cancer risk. Br J Cancer. 2011;104:6–11.
36 Boffetta P, Couto E, Wichmann J, et al. Fruit and vegetable intake and overall cancer risk in the European Prospective Investigation into Cancer and Nutrition (EPIC). J Natl Cancer Inst. 2010;102:529–37.
37 Ioannidis JPA, Siontis GCM. Re: Fruit and vegetable intake and overall cancer risk in the European Prospective Investigation into Cancer and Nutrition. J Natl Cancer Inst. 2011;103:279.
38 Hung H-C, Joshipura KJ, Jiang R, et al. Fruit and vegetable intake and risk of major chronic disease. J Natl Cancer Inst. 2004;96:1577–84.
39 Takachi R, Inoue M, Ishihara J, et al. Fruit and vegetable intake and risk of total cancer and cardiovascular disease: Japan Public Health Center-Based Prospective Study. Am J Epidemiol. 2008;167:59–70.
40 George SM, Park Y, Leitzmann MF, et al. Fruit and vegetable intake and risk of cancer: a prospective cohort study. The American Journal of Clinical Nutrition. 2009;89:347–53.
41 Lin J, Cook NR, Albert C, et al. Vitamins C and E and beta carotene supplementation and cancer risk: a randomized controlled trial. J Natl Cancer Inst. 2009;101:14–23.
42 Bjelakovic G, Nikolova D, Simonetti RG, Gluud C. Antioxidant supplements for preventing gastrointestinal cancers. Cochrane Database Syst Rev 2008; CD004183.
43 Ng K, Meyerhardt JA, Chan JA, et al. Multivitamin use is not associated with cancer recurrence or survival in patients with stage III colon cancer: findings from CALGB 89803. J Clin Oncol. 2010;28:4354–63.
44 Druesne-Pecollo N, Latino-Martel P, Norat T, et al. Beta-carotene supplementation and cancer risk: a systematic review and metaanalysis of randomized controlled trials. International journal of cancer Journal international du cancer 2010;127:172–84.
45 Giovannucci E, Stampfer MJ, Colditz GA, et al. Multivitamin use, folate, and colon cancer in women in the Nurses’ Health Study. Ann Intern Med. 1998;129:517–24.
46 Larsson SC, Giovannucci E, Wolk A. A prospective study of dietary folate intake and risk of colorectal cancer: modification by caffeine intake and cigarette smoking. Cancer Epidemiol Biomarkers Prev. 2005;14:740–3.
47 Sanjoaquin MA, Allen N, Couto E, Roddam AW, Key TJ. Folate intake and colorectal cancer risk: a meta-analytical approach. International journal of cancer Journal international du cancer 2005;113:825–8.
48 Collin SM, Metcalfe C, Refsum H, et al. Circulating folate, vitamin B12, homocysteine, vitamin B12 transport proteins, and risk of prostate cancer: a case-control study, systematic review, and meta-analysis. Cancer Epidemiol Biomarkers Prev. 2010;19:1632–42.
49 Lippman SM, Klein EA, Goodman PJ, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA. 2009;301:39–51.
50 Gandini S, Boniol M, Haukka J, et al. Meta-analysis of observational studies of serum 25-hydroxyvitamin D levels and colorectal, breast and prostate cancer and colorectal adenoma. International journal of cancer Journal international du cancer. 2011;128:1414–24.
51 Giovannucci E, Chan AT. Role of vitamin and mineral supplementation and aspirin use in cancer survivors. J Clin Oncol. 2010;28:4081–5.
52 Manson JE, Mayne ST, Clinton SK. Vitamin D and prevention of cancer – ready for prime time? N Engl J Med. 2011;364:1385–7.
53 Tang JY, Fu T, Leblanc E, et al. Calcium Plus Vitamin D Supplementation and the Risk of Nonmelanoma and Melanoma Skin Cancer: Post Hoc Analyses of the Women’s Health Initiative Randomized Controlled Trial. J Clin Oncol 2011;
54 Yin L, Grandi N, Raum E, Haug U, Arndt V, Brenner H. Meta-analysis: Serum vitamin D and breast cancer risk. European Journal of Cancer. 2010;46:2196–205.
55 Schütze M, Boeing H, Pischon T, et al. Alcohol attributable burden of incidence of cancer in eight European countries based on results from prospective cohort study. BMJ. 2011;342:d1584.
56 Morgan TR, Mandayam S, Jamal MM. Alcohol and hepatocellular carcinoma. Gastroenterology. 2004;127:S87–96.
57 Goldstein BY, Chang S-C, Hashibe M, Vecchia CL, Zhang Z-F. Alcohol consumption and cancers of the oral cavity and pharynx from 1988 to 2009: an update. Eur J Cancer Prev. 2010;19:431–65.
58 Islami F, Fedirko V, Tramacere I, et al. Alcohol drinking and esophageal squamous cell carcinoma with focus on light-drinkers and never-smokers – A systematic review and meta-analysis. International journal of cancer Journal international du cancer 2010;
59 Fedirko V, Tramacere I, Bagnardi V, et al. Alcohol drinking and colorectal cancer risk: an overall and dose-response meta-analysis of published studies. Ann Oncol 2011;
60 Li CI, Chlebowski RT, Freiberg M, et al. Alcohol consumption and risk of postmenopausal breast cancer by subtype: the women’s health initiative observational study. J Natl Cancer Inst. 2010;102:1422–31.
61 Kwan ML, Kushi LH, Weltzien E, et al. Alcohol consumption and breast cancer recurrence and survival among women with early-stage breast cancer: the life after cancer epidemiology study. J Clin Oncol. 2010;28:4410–6.
62 Rizos C, Papassava M, Golias C, Charalabopoulos K. Alcohol consumption and prostate cancer: a mini review. Exp Oncol. 2010;32:66–70.
63 Khan N, Mukhtar H. Cancer and metastasis: prevention and treatment by green tea. Cancer Metastasis Rev. 2010;29:435–45.
64 Li Y, Chang S-C, Goldstein BY, et al. Green tea consumption, inflammation and the risk of primary hepatocellular carcinoma in a Chinese population. Cancer epidemiology 2011;
65 Boehm K, Borrelli F, Ernst E, et al. Green tea (Camellia sinensis) for the prevention of cancer. Cochrane Database of Systematic Reviews (Online) 2009; CD005004.
66 Grove KA, Lambert JD. Laboratory, epidemiological, and human intervention studies show that tea (Camellia sinensis) may be useful in the prevention of obesity. J Nutr. 2010;140:446–53.
67 Bravi F, Bosetti C, Tavani A, et al. Coffee drinking and hepatocellular carcinoma risk: a meta-analysis. Hepatology. 2007;46:430–5.
68 Larsson SC, Wolk A. Coffee consumption and risk of liver cancer: a meta-analysis. Gastroenterology. 2007;132:1740–5.
69 Freedman ND, Curto TM, Lindsay KL, et al. Coffee Consumption is Associated with Response to Peginterferon and Ribavirin Therapy in Patients with Chronic Hepatitis C. Gastroenterology 2011;
70 Zhang X, Albanes D, Beeson WL, et al. Risk of colon cancer and coffee, tea, and sugar-sweetened soft drink intake: pooled analysis of prospective cohort studies. J Natl Cancer Inst. 2010;102:771–83.
71 Aleksandrova K, Boeing H, Jenab M, et al. Metabolic Syndrome and Risks of Colon and Rectal Cancer: the European Prospective Investigation into Cancer and Nutrition Study. Cancer Prev Res (Phila) 2011;
72 Renehan AG, Tyson M, Egger M, Heller RF, Zwahlen M. Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet. 2008;371:569–78.
73 Welzel TM, Graubard BI, Zeuzem S, El-Serag HB, Davila JA, McGlynn KA. Metabolic syndrome increases the risk of primary liver cancer in the United States: A study in the SEER-medicare database. Hepatology 2011;
74 Campbell PT, Jacobs ET, Ulrich CM, et al. Case-control study of overweight, obesity, and colorectal cancer risk, overall and by tumor microsatellite instability status. J Natl Cancer Inst. 2010;102:391–400.
75 Nguyen PL, Ma J, Chavarro JE, et al. Fatty acid synthase polymorphisms, tumor expression, body mass index, prostate cancer risk, and survival. J Clin Oncol. 2010;28:3958–64.
76 Ewertz M, Jensen M-B, Gunnarsdóttir K, et al. Effect of obesity on prognosis after early-stage breast cancer. J Clin Oncol. 2011;29:25–31.
77 Erickson K, Patterson RE, Flatt SW, et al. Clinically defined type 2 diabetes mellitus and prognosis in early-stage breast cancer. J Clin Oncol. 2011;29:54–60.
78 Peairs KS, Barone BB, Snyder CF, et al. Diabetes mellitus and breast cancer outcomes: a systematic review and meta-analysis. J Clin Oncol. 2011;29:40–6.
79 Basen-Engquist K, Chang M. Obesity and cancer risk: recent review and evidence. Curr Oncol Rep. 2011;13:71–6.
80 Pischon T, Boeing H, Hoffmann K, et al. General and abdominal adiposity and risk of death in Europe. N Engl J Med. 2008;359:2105–20.
81 Martin B, Ji S, Maudsley S, Mattson MP. “Control” laboratory rodents are metabolically morbid: why it matters. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:6127–33.
82 Heilbronn LK, Ravussin E. Calorie restriction and aging: review of the literature and implications for studies in humans. Am J Clin Nutr. 2003;78:361–9.
83 Hursting SD, Lavigne JA, Berrigan D, Perkins SN, Barrett JC. Calorie restriction, aging, and cancer prevention: mechanisms of action and applicability to humans. Annu Rev Med. 2003;54:131–52.
84 Martin B, Golden E, Egan JM, Mattson MP, Maudsley S. Reduced energy intake: the secret to a long and healthy life? IBS J Sci. 2007;2:35–9.
85 Kagawa Y. Impact of Westernization on the nutrition of Japanese: changes in physique, cancer, longevity and centenarians. Prev Med. 1978;7:205–17.
86 Hughes LAE, van dB, Piet A, de B, Adriaan P, et al. Early life exposure to famine and colorectal cancer risk: a role for epigenetic mechanisms. PLoS One 2009;4:e7951.
87 Benedict FG, Miles WR, Roth P, Smith HM. Human Vitality and Efficiency under Prolonged Restriction Diet. Washington: Carnegie Institution of Washington, 1919.
88 Dirks AJ, Leeuwenburgh C. Caloric restriction in humans: potential pitfalls and health concerns. Mech Ageing Dev. 2006;127:1–7.
89 Mattson MP, Cutler RG, Camandola S. Energy intake and amyotrophic lateral sclerosis. Neuromolecular Med. 2007;9:17–20.
90 Mattson MP. Energy intake, meal frequency, and health: a neurobiological perspective. Annu Rev Nutr. 2005;25:237–60.
91 Rocha NS, Barbisan LF, de O, Maria Luiza Cotrim, de C, João Lauro Viana. Effects of fasting and intermittent fasting on rat hepatocarcinogenesis induced by diethylnitrosamine. Teratog, Carcinog Mutagen. 2002;22:129–38.
92 Berrigan D, Perkins SN, Haines DC, Hursting SD. Adult-onset calorie restriction and fasting delay spontaneous tumorigenesis in p53-deficient mice. Carcinogenesis. 2002;23:817–22.
93 Popkin BM, Duffey KJ. Does hunger and satiety drive eating anymore? Increasing eating occasions and decreasing time between eating occasions in the United States. Am J Clin Nutr. 2010;91:1342–7.
94 Mattson MP. The need for controlled studies of the effects of meal frequency on health. Lancet. 2005;365:1978–80.
95 Maalouf M, Rho JM, Mattson MP. The neuroprotective properties of calorie restriction, the ketogenic diet, and ketone bodies. Brain Res Rev. 2009;59:293–315.
96 Nebeling LC, Miraldi F, Shurin SB, Lerner E. Effects of a ketogenic diet on tumor metabolism and nutritional status in pediatric oncology patients: two case reports. J Am Coll Nutr. 1995;14:202–8.
97 Zuccoli G, Marcello N, Pisanello A, et al. Metabolic management of glioblastoma multiforme using standard therapy together with a restricted ketogenic diet: Case Report. Nutr Metab. (Lond) 2010;7:33.
98 Mavropoulos JC, Isaacs WB, Pizzo SV, Freedland SJ. Is there a role for a low-carbohydrate ketogenic diet in the management of prostate cancer? Urology. 2006;68:15–8.
99 Marantz PR, Bird ED, Alderman MH. A call for higher standards of evidence for dietary guidelines. Am J Prev Med. 2008;34:234–40.