Chronic liver inflammation and hepatocellular carcinoma: persistence matters
DOI: https://doi.org/10.4414/smw.2011.13197
YT
Boege, F
Reisinger, M
Heikenwalder
Summary
Inflammatory responses in the liver – a central constituent of hepatic wound healing – can be self-limited or persistent depending on the aetiology, liver health state, concentration of toxins or pathogens, and the time frame of exposure to toxins or infection. In case the immune system eradicates a pathogen or in case toxin-exposure is transient, acute hepatitis resolves and the affected liver tissue regenerates ad integrum. However, in many cases liver damage remains chronic. Irrespective of the aetiology, chronic liver damage drives chronic hepatitis and hepatocyte death as well as compensatory proliferation, reflecting liver regeneration. Over time this potentially promotes further hepatic damage, fibrosis, cirrhosis and liver cancer. Here, we review the current knowledge on how chronic liver injury and inflammation is triggered and maintained, and how inflammation is linked to liver cancer. We also discuss the most frequently used animal models for damage or inflammation induced liver cancer and their suitability for conducting clinically relevant research.
Summary
Hepatitis can be elicited in response to a plethora of diverse insults to the liver. Chronic inflammation is associated with persistent liver damage and consecutive regeneration, potentially leading to fibrosis and cirrhosis and the development of HCC. Human HCC, even of the same aetiology, reveal a broad clinical, morphological and molecular spectrum, however, they generally have a bad prognosis. The efficiency of drugs is currently limited, but might be improved by the identification of specific molecular targets. Several mouse models are available which are used to recapitulate different aetiologies of human hepatocarcinogenesis. It is now of paramount importance to determine how these models are transferable to human HCC and how they can be thus exploited for interventional studies.
Acknowledgments: We thank Jay Tracy, Barbara Zadnich, Sukumar Namineni, Dr. Nicole Simonavicius and Dr. Barbara Stecher for reading the manuscript and for valuable input. We are thankful to all members of our laboratory for discussions and apologise to those authors whose contributions were not cited due to space limitations.
Acute and chronic hepatitis – inflammation of the liver
The term hepatitis (i.e. inflammation of the liver) designates a broad spectrum of diseases. The clinical course of hepatitis ranges from no or only limited symptoms, clinical appearance characterised by poor appetite, malaise and jaundice to fulminant liver failure [1]. Hepatitis can be self-limited or persistent. The terms acute and chronic hepatitis are defined by the duration of disease with less or more than six months, respectively. The spectrum of aetiologies causing hepatitis is broad and comprises of infectious agents, mainly hepatotropic but also other viruses [2–4], bacteria, protozoa, autoimmunity, chronic cholestatic diseases like autoimmune hepatitis (AIH) [5], primary biliary cirrhosis (PBC) [6] and primary sclerosing cholangitis (PSC), metabolic diseases (e.g. Wilson’s disease), a plethora of nutritive-toxic insults including alcohol abuse [7], non-alcoholic fatty liver disease (NAFLD), and various drugs [8]. A common feature of hepatitis is the presence of inflammatory cells and damage of liver tissue.
Characteristic histological features of acute hepatitis include inflammatory cells in portal tracts and within the parenchyma, focal or zonal necrosis, dying hepatocytes (acidophilic bodies, apoptotic cells), resulting in architectural disarray and cholestasis [1]. With disease progression, features of resolution become visible including the appearance of pigment-laden macrophages and signs of regeneration. The degree to which the individual features can be observed varies and depends on the underlying disease and clinical setting.
Chronic hepatitis is characterised by the persistence of inflammatory cells and the destruction of liver cells. Again, morphologic patterns among chronic hepatitis of diverse aetiologies are not uniform, but pathognomonic or characteristic features exist. For example, the presence of so-called ground glass hepatocytes (i.e., granular eosinophilic) staining due to abundant hepatitis B antigens (HBsAg) in the endoplasmic reticulum of hepatocytes indicates chronic hepatitis B infection (figure 1A). The presence of inflammatory activity in the interphase between portal tract and lobules with numerous plasma cells is characteristic of AIH (figure 1B).
Remarkably, the liver has a unique regenerative capacity and can replace a significant loss of liver cells by starting a programme of compensatory proliferation, driving hepatocytes from a resting G0 to a replicative G1 state [9]. However, chronic liver damage and regeneration results in “scarring” of the liver: A typical sequela of chronic hepatitis is the development of liver fibrosis (i.e., the deposition of excessive fibrous tissue [10]). The location of fibrosis also varies depending on the aetiology and duration of liver disease. Fibrosis may proceed such that normal lobules are replaced by architecturally abnormal nodules, resulting mainly from regenerative hyperplasia, separated by fibrous tissue. At this point, the late stage of progressive fibrosis, called cirrhosis, has developed which is generally regarded as irreversible. Patients with cirrhosis are prone to a variety of complications and have a reduced life expectancy. Decompensated cirrhosis is characterised by life-threatening complications. Another complication of cirrhosis is the development of hepatocellular carcinoma (HCC). HCC develop in the background of cirrhosis of almost any cause, including AIH [11] and PBC [12]. In contrast to a hepatitis B carrier state, cirrhotic transformation of the liver seems to be required for development of HCC in both AIH and PBC.
Chronic inflammation – a state which fosters multiple hallmark functions of cancer
When Rudolph Virchow presented his concept of tumour development in a lecture series on “Die krankhaften Geschwülste”, he postulated a link between wound healing, scar formation, inflammation and the development of tumours [13, 14]. His speculation on the existence of a cancer-bacillus was even more remarkable at that time, especially as we know today that he was correct [14, 15]. In the last 25 years, numerous clinical and epidemiological studies have corroborated the link between chronic inflammation and carcinogenesis [16]. These studies have demonstrated without any doubt that chronic inflammation promotes the risk for cancer development. Consequently, the “inflammatory microenvironment” was added as the seventh hallmark of cancer complementing the list of hallmarks published in a seminal review in 2000 [15, 17, 18] which included: (1) sustained proliferative signalling, (2) evading growth suppressors, (3) resisting cell death, (4) enabling replicative immortality, (5) inducing angiogenesis, and (6) activating invasion and metastasis [15]. Recently, this list was updated by adding reprogramming of energy metabolism and evading immune destruction as further conceptual hallmarks [19].
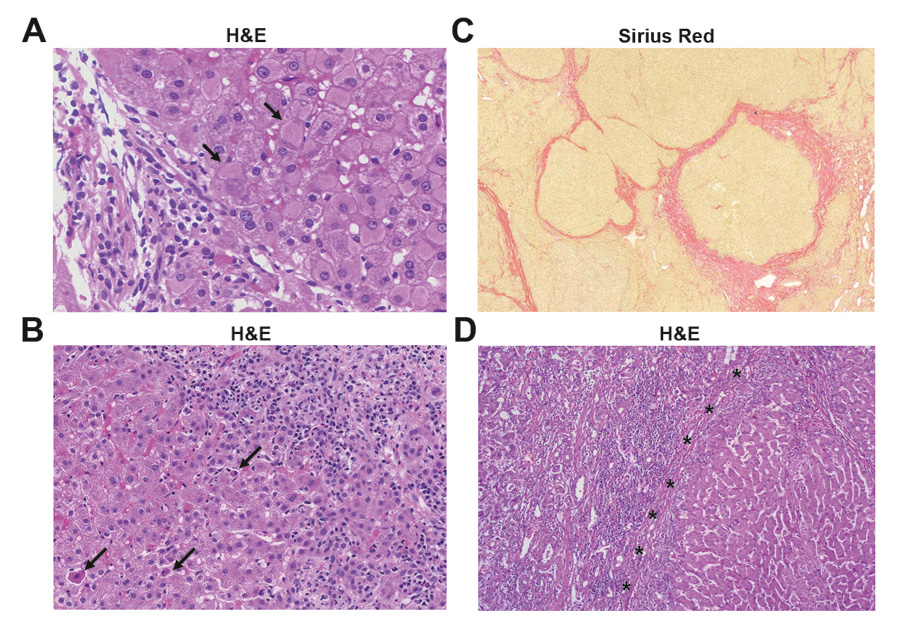
Figure 1
Histology of various human liver diseases. (A) Histology of chronic hepatitis B infection. Chronic inflammation of mild activity mainly confined to the portal tract (left) and numerous hepatocytes with ground class cytoplasm (arrows) due to accumulation of viral proteins (HBsAg) (haematoxylin and eosion staining). (B) Histology of autoimmune hepatitis (AIH). Histology reveals portal and interphase inflammatory activity and numerous plasma cells (right). Note the presence of several apoptotic hepatocytes (arrows) (haematoxylin and eosion staining). (C) Histology of cirrhotic liver tissue. Cirrhosis is characterised by architecturally abnormal nodules of liver tissue, separated by fibrous tissue (red, elastica van Gieson staining) (D) Histology of hepatocellular carcinoma (left) adjacent to cirrhotic liver tissue (right). Note the inflammatory infiltrate at the border between the tumour and surrounding tissue (haematoxylin and eosion staining).
Chronic inflammation can be induced by viruses, for example hepatitis B and C viruses (HBV, HCV), which further trigger HCC development [20]. Infection with pathogenic bacteria (e.g., Helicobacter pylori) promotes gastritis and increases the risk for gastric cancer and mucosa-associated lymphoid tissue (MALT) lymphoma [21]. Infection with parasites (e.g. Schistosoma) causes chronic inflammation potentially leading to bladder, liver or colorectal cancer [22]. Moreover, as noted above, autoimmune diseases can trigger cancer development: Autoimmune hepatitis increases the risk for HCC development [23], and inflammatory bowel diseases (e.g., ulcerative colitis, Crohn’s disease) are a predisposition to colon cancer formation [24]. Besides, PSC can lead to cholangiocellular carcinoma (CCC) [25]. In addition to infectious and autoimmune disorders, chronic or acute intoxication (e.g., alcohol, drug abuse, aflatoxin-b) can cause inflammation-induced carcinogenesis [26]. Only recently dietary and genetic obesity were also shown to promote hepatitis and tumourigenesis [27, 28].
However, numerous clinical reports and experimental data support the idea that inflammation (acute or chronic) can also be anti-carcinogenic [29-32]. This was already noted by the oncologist William Coley who used concomitant activation of the adaptive immune system (e.g., by injection of dead bacteria Streptococcus pyogenes, Serratia marcescens and active endotoxins) to treat cancer patients [33]. Most likely the hereby induced inflammation activates the innate and adaptive immune cells to trigger destruction of tumour cells. Today this therapeutic concept is used to treat bladder cancer [34].
Although immense progress has been made in the past to understand the mechanism(s) by which chronic inflammation influences tissue integrity, chromosomal stability, apoptosis, proliferation and cancer development, the exact pathways and cellular ingredients that define inflammation as anti- or pro-carcinogenic remain unknown. The initial composition of immune cells in the target organ, the cells infiltrating or surrounding tumour tissue, the interaction of the tumour stroma with immune cells, the expression of cytokines and chemokines and the organ in which chronic inflammation occurs might explain why inflammation is a double- edged sword [15, 19, 29, 35]. Furthermore, it remains elusive if the transition from chronic inflammation to cancer underlies specific pathways or if the existence of an inflammatory environment unspecifically increases the stochastic likelihood to cause cancer. Presumably, both scenarios are possible depending on various genetic host factors and the aetiology driving cancer.
Hepatocellular carcinoma (HCC)
Today, HCC is the third most common cause for cancer-related death world-wide [36]. In some African and Asian countries HCC has become the most prevalent cause for cancer-related death [37]. Given the effort invested in the past, the clinical success to pharmacologically treat HCC patients has been limited. Currently, liver transplantation is the most effective way to prolong the life of HCC patients [38]. However, new drugs are being generated and investigated. Recently, various inhibitor molecules have entered clinical practice. As such, Sorafenib (Nexavar®) is one of the new therapeutic agents inhibiting pro-angiogenic and tumourigenic receptor tyrosine kinases (e.g., c-Kit; VEGFR-1, -2, -3; PDGFR-β). Its efficacy in the context of HCC treatment was demonstrated in two large phase III clinical trials [39, 40]. In addition, other therapeutics, such as Regorafenib (BAY 73-4506), are being investigated for their potential as anti-tumour regimens.
Although these regimens successfully prolong patient survival, their effect is limited and treatment is rather palliative [39, 40]. Furthermore, since HCC do not resemble a single entity but rather a diverse spectrum of cancers, various therapeutic approaches might be needed to treat HCC subtypes. Due to the outlined problems, a systemic therapeutic approach to treat liver cancer is not available, underlining the need for new targets to be investigated in basic and clinical research.
The most common cause for chronic hepatic inflammation in humans is infection with HBV or HCV [41]. The spread of HBV and HCV has resulted in approximately 500 million people with persistent virus infections [41], some of which will lead to liver cancer causing the strong rise in HCC incidence. At the same time, eradication of these chronic viral infections has turned out to be very ineffective [42].
Therefore, analysis and identification of fundamental inflammatory signalling pathways causing the transition from chronic liver injury to dysplasia and HCC could depict a plethora of new targets or predictive markers to identify and treat patients with chronic liver inflammation. Of highest importance will be the identification of pathways that stabilise liver tumourigenesis, as not all HCC depend on an inflammatory environment and therefore may not respond to an anti-inflammatory therapy.
From mice to men: A difficult task
In light of the above information, modelling liver cancer in animals appears to be crucial especially to determine which mouse models exist and what human pathology they reflect. Indeed, establishment of mouse models that mimic particular pathological aspects of liver tumourigenesis in humans can “only” be the initial step. It should be followed by a detailed analysis of the pathways that the particular model relies on. Furthermore, knowledge of which specific pathological features of human HCC subtypes the mouse model represents may increase the efficacy of clinical trials in the future.
The link between chronic inflammation preceding fibrosis and liver cancer has been recapitulated in rodent models [19, 43]. In the last 20 years, a huge amount of rodent models of chronic or acute liver damage-induced carcinogenesis (e.g., chemically, metabolically or genetically) have been established. Some of the most important ones are discussed in this review (see also table 1).
|
Table 1Frequently used HCC mouse models. Many different mouse models have been established in the past 30 years. Some of the most frequently used are listed below. Due to space limitations we were not able to list all HCC models available. |
|
Models
|
Phenotype
|
Known mechanism
|
References
|
| Transgenic models |
|
|
|
| AlbLTαβ |
Chronic inflammation, fibrosis, dysplasia, HCC at 12 mo |
Chronic hepatitis, cell damage, apoptosis, compensatory hepatocyte proliferation, genomic instability |
[53] |
| Alb-c-myc |
Dysplasia, preneoplastic foci, adenoma, HCC at 12–15 mo |
Apoptosis induction, enhanced proliferation, genomic instability |
[73] |
| c-myc/TGFα |
Dysplastic and apoptotic changes in hepatocytes multiple focal lesions, HCC at 8 mo |
Acceleration of neoplastic development compared to single- transgenic models |
[69, 74] |
| MT-TGF-α |
Higher DNA content, dysplastic hepatocytes, adenmoas and HCC at 12 mo |
Mitogenic effect due to chronic exposure to high levels of TNFα |
[75, 76] |
| Transgenic models / viral genes |
|
|
|
| c-myc/HBX |
Expansion of hepatic lobules, dysplastic foci, neoplastic nodules, HCC at 6–8 mo |
2-fold higher hepatocyte proliferation rate compared to c-myc-only transgenic mice |
[71, 77] |
| HBV large envelope protein |
Inflammation, regenerative hyperplasia, aneuploidy, HCC at 15 mo |
Accumulation of HBsAg in the ER leading to hepatocyte death |
[78] |
| HBV X protein |
Cytoplasmic vacuolations, neoplastic lesions, HCC at 13 mo |
Transcriptional transactivation of viral genes leading to alter host gene expression and carcinomas |
[79] |
| HCV core |
Steatosis, HCC at 16 mo |
Core protein acts as transcriptional activator affecting the proliferative ability of hepatocytes, ROS production, transcriptional activation of TNF and IL1 |
[80] |
| SV40 T-Ag |
Hyperplastic hepatocytes, dysplasia, HCC at 3 mo, lung metastasis |
SV40 T-Ag-mediated immortalisation, cell transformation, aberrant DNA replication |
[81, 82] |
| TgAlb-1/HBV |
Inflammatory infiltrates, focal hepatocellular necrosis, disorganisation of lobular architecture, HCC at 17 mo |
Expression of HBsAg in hepatocytes – HBsAg-tolerant mice – HCC formation due to CTL mediated cell damage |
[44] |
| Knockout models |
|
|
|
| Dicer1Δhep
|
Depletion of glycogen storage, steatosis, HCC at 12 mo |
Impaired lipid and glucose metabolism, increased expression of cell cycle promoting genes, increased proliferation and apoptosis |
[72] |
| MCL-1Δhep
|
Apoptoses, architectural disarray, fibrosis, dysplastic nodules and HCC at 8 mo |
Spontaneous hepatocyte apoptosis leading to chronic liver damage and compensatory proliferation |
[60, 61] |
| MCL-1Δhep/p53-/-
|
More severe liver damage and higher tumour incidence compared to MCL-1Dhep |
p53 inhibits apoptotic effectors and is known to interact with MCL-1 |
[62] |
| Mdr2-/-
|
Cholastasis, inflammation, dysplasia, dysplastic nodules, HCC at 10 mo, metastasis |
Mice are unable to secrete phospholipids into bile leading to liver disease |
[49, 83] |
| Mst1/2-/-
|
Loss of hepatocyte quiescence leading to overgrowthHCC at 3 mo |
Mst1/2 deficiency leads to resistance to Fas-induced apoptosis and loss of Yap1 phosporylation |
[65] |
| NEMOLPC-KO
|
Steatohepatitis, liver fibrosis, dysplasia, HCC at 11 mo |
Increased apoptotic hepatocyte death and subsequent compensatory proliferation |
[58] |
| TAK1LPC-KO
|
Steatohepatitis, liver fibrosis, dysplasia, biliary cirrhosis, HCC at 4 mo |
TAK1 deficiency leads to caspase-3- mediated apoptosis accompanied by enhanced liver cell proliferation |
[59] |
| Chemically-induced models |
|
|
|
| Aflatoxin B1 |
HCC at 11 mo – incidence rate depends on the mouse strain |
Chromosomal aberrations, sister chromatide exchange, chromosomal strand breaks, uncontrolled DNA synthesis |
[84, 85] |
| Choline deficient diet |
Steatosis, HCC at 11–12 mo |
Formation of oval cells due to oxidative DNA damage and chromosomal instability finally leading to generation of hepatocytes |
[86] |
| DEN wild-typeDEN IKKβΔhep
|
Acute hepatitis, hepatocyte proliferation, HCC at 8–10 mo, metastasis3-fold higher tumour number compared to wt |
Chemically induced DNA damage leading to genetic mutationsIncreased ROS production and JNK activation, augmented hepatocyte turn-over compared to wt |
[51] |
| Transplantation models |
|
|
|
| Metastatic HCC xenografts(LCI-D20) |
100% lung metastasis incidence after 15 days |
Orthotopic implantation of metastatic tumour tissues |
[87] |
The TgAlb-1, HBV mouse model
In an elegant study, the pathogenetic mechanisms responsible for HCC formation during chronic HBV infection were investigated [44]. Using transgenic mice which expressed the HBV surface antigen (HBsAg) but which were immunologically tolerant to HBsAg at the T-cell level, the authors investigated how a chronic immune response against a viral antigen drives inflammation and HCC formation [44]. TgAlb-1, HBV mice, which were thymectomised, irradiated, bone marrow-reconstituted and adoptively transferred with HBsAg-specific CTLs, developed a severe MHC class I-restricted necroinflammatory liver disease. Importantly, it was shown that this chronic immunemediated liver cell injury triggers HCC development even in the absence of viral transactivation, insertional mutagenesis, and genotoxic chemicals [44]. This model shows that chronic inflammation induces liver cancer, thereby recapitulating one important aspect of virus-induced chronic hepatitis and liver cancer formation in humans. Some of the most important mouse models for HBV- or HCV- induced liver cancer formation and their underlying mechanisms are listed in table 1.
Chemically induced HCC – the DEN mouse model
The most intensively studied model of chemically induced liver cancer development is the use of diethylnitrosamine (DEN), both in rats and mice [32, 45]. In mice, a single injection of DEN in 2-week-old animals leads to acute hepatitis, a pronounced Kupffer cell response, hepatocyte proliferation and DNA damage, finally leading to HCC at approximately 8 to 10 months of age [32]. Treatment with DEN has several advantages: (a) DEN can be used in mice of various genetic backgrounds, (b) DEN treatment induces high HCC incidence, and (c) it is highly reproducible. In addition, HCC in DEN- treated mice recapitulates one important feature of carcinogenesis: metastasis. However, the major event in DEN-induced hepatocarcinogenesis is the induction of mutations and a strong pro-proliferative environment in a previously healthy liver. Although this occurs concomitantly with acute liver damage, the sequence of primary DNA damage followed by inflammation and Kupffer cell activation does not necessarily mimic the chain of events of an inflammation-induced paradigm. Nevertheless, DEN treatment has depicted important molecular and cellular pathways involved in HCC development (e.g., Stat-3P; IL6) and can be used to identify new targets in order to block chemically induced liver carcinogenesis in humans [32, 46].
Additionally to DEN, several other chemical carcinogens such as a aflatoxin B1 (AFB) or choline-deficient diet have been used to initiate HCC development in mice (table 1). AFB is of particular interest because of its contribution to the high rates of HCC in China and Western Africa [47, 48].
The mdr2-ko mouse model
The mdr2-knockout mouse model embodies the prototype of a defined genetic modification leading to chronic inflammation and liver cancer. It is used to classify the pathways involved in chronic inflammation-induced HCC formation [49]. Deficiency of the multi-drug resistance gene 2 (mdr2), a biliary transporter protein, causes cholestatic hepatitis and liver cancer [49]. Mdr2 deficiency induces different subsequent phases: cholestasis, inflammation, dysplasia, dysplastic nodules, carcinoma and metastasis, recapitulating aspects of inflammation-induced carcinogenesis in humans. The cholestatic and inflammatory state triggers hepatocyte-derived NF-kb through TNF up-regulation in adjacent endothelial and inflammatory cells [49]. Turning off NF-κb in mdr2-ko hepatocytes did not affect liver cancer development nor early phases of hepatocyte transformation [49]. NF-κb inhibition by anti-TNF treatment – even at the late stage of tumourigenesis – resulted in apoptosis of transformed hepatocytes and blocked hepatocarcinogenesis. Therefore, NF-κb signalling might be a pro-carcinogenic factor representing a possible target for anti-tumour therapy.
The NF-κB pathway controls transcription of inflammatory, pro-proliferative and anti-apoptotic genes [29] and is one of the most important and best-studied inflammatory signalling cascades [50]. Activation of NF-κB is documented by phosphorylation of the I-kappa-B kinase (IKK) complex, consisting of two catalytic subunits, IKKα and IKKβ, and a regulatory subunit called IKKγ. Upon recruitment of adaptor proteins (e.g., TRAF2; RIP1) and activation by the TGFβ-activated kinase 1 (TAK1), phosphorylation of the inhibitory protein IκBα occurs and nuclear translocation of NF-kB is initiated [31, 43]. However, NF-κB signalling is complex, as exemplified by the DEN model: Hepatocyte-specific deletion of IKKβ leads to increased HCC formation [51], suggesting an anti-tumourigenic role of IKKβ – which is in contrast to data from mdr2-ko mice. However, whether or not depletion of IKKβ alone changes the behaviour of the remaining IKK complex to solely drive carcinogenesis remains elusive.
The AlbLTαβ mouse model
Lymphotoxin (LT) acts directly on hepatocytes which express high levels of LTβR but little LT [52, 53]. T-cell-derived LT and LIGHT signalling to hepatocytes controls lipoprotein homoeostasis [54]. Moreover, LTbR signalling is important for liver regeneration through T-cell-derived LT expression [55] and LTβR signalling regulates hepatic stellate cell function and wound healing [56].
We recently showed that LTα, β and their receptor (LTbR) are increased in intrahepatic lymphocytes and hepatocytes of patients with HBV- or HCV-induced chronic hepatitis or HCC [53]. Various other liver diseases of non-viral aetiology have shown significantly lower levels of LTα, β and LTβR mRNA expression [35, 53].
Thus, the question remained whether LT expression in mice suffices to induce liver inflammation and HCC. Indeed, transgenic expression of LT specifically in the liver recapitulated some features of human pathology seen in patients with chronic hepatitis B or C. AlbLTαβ mice developed hepatitis and HCC (figure 2). Furthermore, suppression of LTβR signalling reduced the incidence of chronic hepatitis and abolished liver cancer development [57].
IKKβ’s role in tumour-promotion was recapitulated in AlbLTαβ mice backcrossed to IKKβΔhep mice [57]. In contrast to the mdr2-ko mouse model, liver tumour formation in AlbLTαβ mice also occurred in the absence of TNFR1 [49, 57].
How can the contradictory role of IKKβ signalling be reconciled, when comparing the DEN, mdr2-ko and AlbLTαβ models in HCC formation? IKKβ signalling is crucial for hepatocytes to respond to and survive acute liver injury and DEN exposure. In its absence, the amount of damaged and proliferative hepatocytes is increased leading to a rise in HCC development. IKKβ signalling enables local chemokine expression by hepatocytes, – as known from humans with chronic hepatitis C –, subsequently leading to chronic inflammation and HCC. Consistently, AlbLTαβ mice that lack B- and T-cells (AlbLTαβ Rag1–/–) also lack chronic hepatitis and HCC [49]. In summary, the role of NF-κB signalling in hepatocarcinogenesis might depend on the model and the type or degree of liver inflammation and injury [29, 32, 50].
The NEMO and the TAK1 mouse model
The role of NF-κB in hepatocarcinogenesis was further investigated by studying NEMO and TAK1 [58, 59]. Both molecules control activation of NF-κB in various cellular systems and mediate inflammatory signalling pathways [32]. Conditional deletion of NEMO or TAK1 in hepatocytes causes spontaneous HCC development; however, liver carcinogenesis was much more aggressive and also coincided with biliary ductopenia and cholestasis in the latter [59]. This suggests that members of the NF-κB signalling pathway possess a tumour preventing capacity. However, the role of NF-κB signalling in HCC formation is probably more complex, taking into account that the early onset of biliary cirrhosis and hepatocarcingenesis in TAK1Δhep mice can be prevented following backcrossing to NEMOΔhep mice [59]. Therefore, NEMO is strongly involved in the highly aggressive phenotype of TAK1Δhep mice in the absence of NF-κb signalling. It is currently unclear if and how these new genetic tumour models recapitulate an entity of human HCC, and future studies are needed [31, 43].
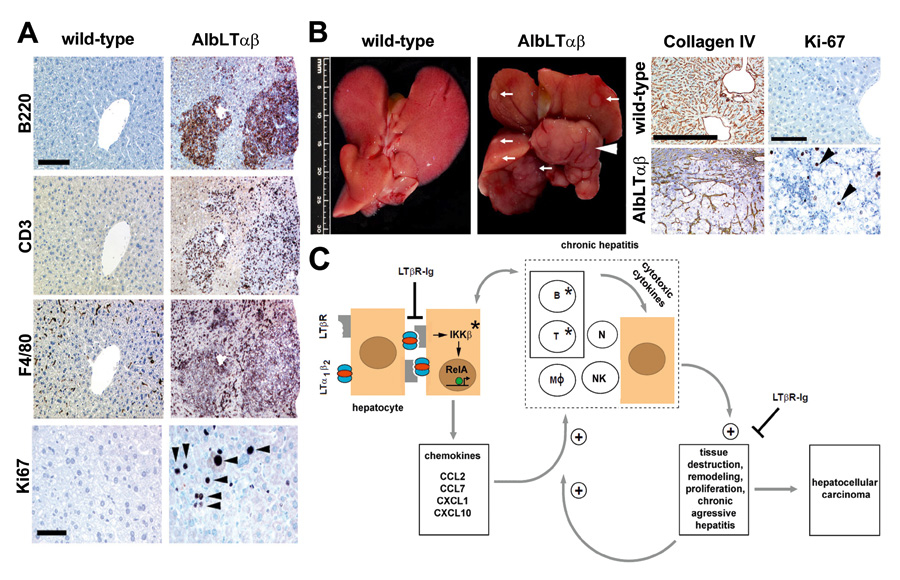
Figure 2
Chronic inflammation-induced liver damage and HCC in AlbLTαβ mice. (A) Immunohistochemistry of representative livers from 9-months-old C57BL/6 and AlbLTαβ mice. B220+ B cells, CD3+ T cells, F4/80+ macrophages, Kupffer cells (scale bar: 150 μm). Ki67+, proliferating hepatocytes (arrowheads) or inflammatory cells are indicated (scale bar: 50 μm). (B, left two panels) Macroscopy of C57BL/6 and AlbLTαβ livers. White arrows indicate tumour nodules. White arrowhead indicates a complete liver lobe affected by HCC. (B, right four panels) Liver histology of 12-month-old C57BL/6 and AlbLTαβ mice. Collagen IV staining highlights the broadening of the liver cell cords, loss of collagen IV networks indicate HCC (scale bar: 200 μm). High numbers of Ki67+proliferating hepatocytes (arrowheads) in AlbLTαβ HCC (scale bar: 100 μm). (C) Scheme of chronic inflammation-induced liver carcinogenesis in AlbLTαβ mice. Hepatocytes (brown) express LTα, LTβ and induce chemokine production (e.g., CCL2, CCL7, CXCL1, CXCL10) in the presence of IKKβ and intrahepatic lymphocytes. Chemoattraction, activation of myeloid cells and lymphocytes expressing chemokine receptors (e.g., CXCR3, CXCR2, CCR2, CCR1) causes hepatitis. Activated, infiltrating immune cells secrete cytotoxic cytokines (e.g., IL6, IL1β, TNFα, IFNγ, LTaβ) causing tissue destruction, hepatocyte proliferation, cell death and tissue remodelling. Tissue destruction and remodelling supports the infiltration of activated inflammatory cells (e.g., myeloid cells) leading to a feed-forward loop towards chronic aggressive hepatitis and HCC. * Genetic depletion of components (IKKb; T and B cells) which leads to a block in chronic hepatitis and HCC development. Blocking LTβR signalling with LTbR-Ig in 9-month-old AlbLTαβ mice reduces chronic hepatitis incidence and prevents HCC. RelA is schematically depicted as a green circle, inducing transcription of NF-κB target genes (arrow). B and T: B and T cells; MØ: macrophages; N: neutrophils; NK: NK cells. Adapted from Haybaeck et al., Cancer Cell, 2009.
A model for apoptosis and proliferation induced HCC: the Mcl-1Δhep mouse
Regulation of tissue homoeostasis is essential for maintaining liver function. A tightly controlled network of pro- and anti-apoptotic factors is crucial to regulate liver tissue development and homoeostasis by sensing extrinsic or intrinsic death signals, subsequently inducing apoptosis to remove damaged hepatocytes and protect cells against malignant transformation. Anti-apoptotic family members (e.g., Bcl-xL, Bcl-2, Mcl-1) interact with pro-apoptotic members (e.g., Bax, Bad) to inhibit the permeabilisation of the mitochondrial membrane. It was found that hepatocyte-specific knock-out of Mcl-1 affected liver tissue homoeostasis and increased the sensitivity of hepatocytes towards apoptosis [60]. Increased apoptosis of hepatocytes in Mcl-1Δhepmice resulted in chronic liver damage, compensatory hepatocyte proliferation, liver fibrosis and finally HCC formation [61] (fig. 3). Tumours revealed a heterogeneous pattern ranging from small dysplastic nodules to HCC. The neoplastic nature of tumours was confirmed by the expression of HCC markers (e.g., gluthamine synthetase) and by chromosomal aberrations [61].
A second study confirmed the link between enhanced hepatocyte apoptosis and tumourigenesis in Mcl-1Δhep mice. Backcrossing of Mcl-1Δhep to p53–/– mice was performed to examine whether the tumour-suppressor p53 plays a role in the induction of spontaneous apoptosis in Mcl-1- deficient hepatocytes [62]. The authors found a high susceptibility of Mcl-1Δhep and p53–/– double knock-out mice (DKO mice) to neonatal death (60%), whereas surviving DKO mice displayed more severe liver damage and a higher tumour incidence. In this model, the loss of p53 promoted liver tumour development due to the multi-faceted role of p53 in apoptosis [62]. p53 is known to bind and inhibit the apoptotic effectors Bak and Bim, as well as to interact with Mcl-1 [63]. Thus, increased liver damage in DKO mice is not surprising when an additional key player in the negative regulation of apoptosis is removed.
In summary, both studies of Mcl-1Δhep mouse models describe the close relationship between a constant loss of liver cells triggered by apoptotic stimuli in correlation with chronic liver injury and spontaneous tumour formation. This sequence recapitulates central aspects of human diseases like chronic hepatitis C [64].
The Mst1/2 knock-out model
In another knock-out mouse model, germline deletions of the protein kinases Mst1 and 2, the mammalian homologues of the Drosophila Hippo kinase, were generated [65]. Loss of function of Hippo accelerates cell cycle progression as shown in Drosophila melanogaster [66, 67]. Zhou et al. showed that Mst1 and Mst2 inhibit the kinase Yes associated protein 1 (Yap1), in order to suppress HCC formation [65]. Remarkably, the Mst-YAP1 pathway is also disrupted in several human HCC [65].
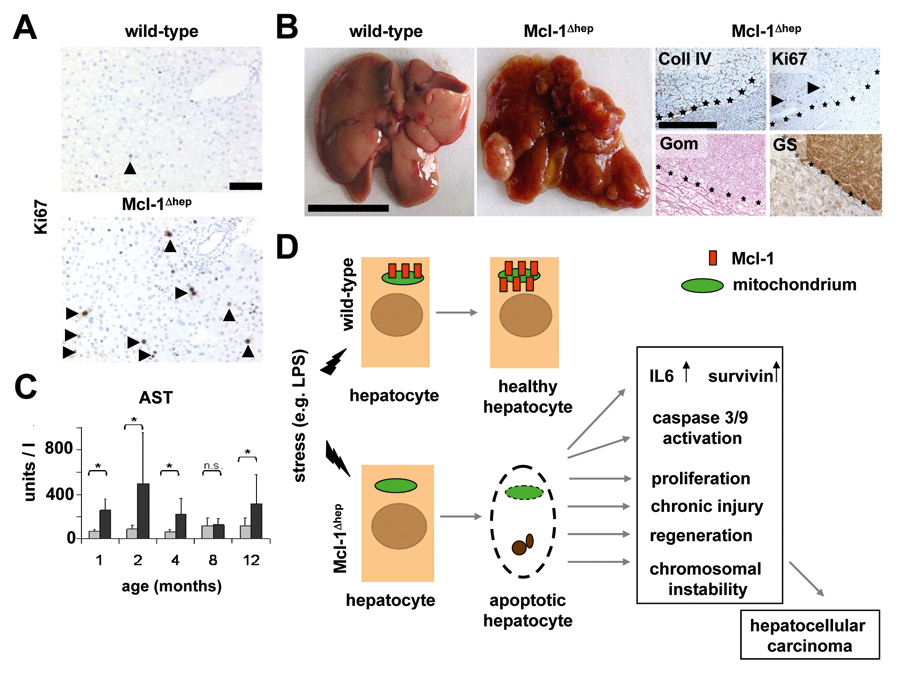
Figure 3
Enhanced proliferation and liver cell damage in Mcl-1Δhep mice. (A) Immunohistochemical analysis of proliferating hepatocytes stained for Ki67. A strong increase in proliferating hepatocytes is found in Mcl-1Δhep mice compared to wild-type mice. (B) Liver damage is assayed by analysis of serum amino transferase levels. In comparison to wild-type mice, Mcl-1Δhep mice display a strong increase in AST and ALT (not shown) levels at early (1–2 months) and later stage (12 months). (C, left two panels) Macroscopic pictures of HCC in Mcl-1Δhep mice. * statistically significant; n.s.: not significant. (C, right four panels) Histological analysis of C57BL/6 and Mcl-1Δhep livers. Collagen IV staining highlights the broadening of the liver cell cords, loss of collagen IV networks indicate HCC (scale bar: 200 μm). High numbers of Ki67+ proliferating hepatocytes, positivity for glutamine syntethase and loss of reticulin fibres (Gomori staining) indicate the presence of HCC. (D) Schematic model explaining the hypersensitivity of Mcl-1Δhep mice to stress-induced apoptosis and its effects on liver homeostasis. Adapted from Weber et al., Hepatology, 2010.
The c-myc/TGFα transgenic mouse model
In 1986 it was shown that hepatitis virus infected woodchucks displayed rearrangement and enhanced expression of the oncogene c-myc in HCC [68]. In addition, the group of Snorri Thorgeirsson demonstrated that co-expression of c-myc and TGFα in mouse livers led to a rise in HCC compared to the expression of either of these transgenes alone [69, 70]. Although morphological similarities were found between the two transgenic lines, the dramatic acceleration, extent and severity of hepatic lesions in c-myc/TGFα mice showed a synergistic effect of c-myc and TGFα [69, 70]. Furthermore, the HBV X gene potentiates c-myc-induced liver oncogenesis in transgenic mice [71]. These transgenic models turned out to be extremely useful to identify oncogene-dependent signalling pathways in human HCC.
Liver-specific Dicer knock-out mice
There is a plethora of publications that indicate that viral infection is correlated with a deregulation of micro-RNAs, and growing evidence suggests that micro-RNAs coordinate various biological processes in the liver [72]. Using a conditional knock-out mouse model with Dicer disrupted specifically in hepatocytes, an enzyme essential for the processing of micro-RNAs, Sekine et al. investigated the consequences of loss of micro-RNAs [72]. Unexpectedly, mice with hepatocytes deficient in Dicer displayed prominent steatosis and depletion of glycogen storage [72]. Dicer elimination resulted in both increased hepatocyte proliferation as well as strong apoptosis. Over time, Dicer-expressing wild-type hepatocytes which had escaped Cre-mediated recombination progressively repopulated the entire liver. Unexpectedly, two thirds of the mutant mice spontaneously developed HCC derived from residual Dicer-deficient hepatocytes at 1 year of age. Therefore, micro-RNAs control liver homoeostasis and HCC formation.
Xenotransplantation of human liver tissue
Xenotransplantation of human liver cancer tissue into immuno-compromised mice made it possible to investigate the efficacy and pharmacokinetics of various antitumor regimens, as well as to investigate signalling mechanisms driving or preventing liver carcinogenesis or metastasis [87]. Tumours can be grafted but can also be established by injection of human cancer cells either subcutaneously (ectopic model) or intrahepatically (orthopic model). Tumour development is rapid and highly reproducible in various xenograft models; however, it is noteworthy that they often show only minor similarities to human tumours.
References
1 Burt A, Portmann B, Ferrell L. MacSween’s pathology of the liver. 2006.
2 Bostan N, Mahmood T. An overview about hepatitis C: a devastating virus. Crit Rev Microbiol. 2010;36(2):91–133.
3 Dienstag JL. Hepatitis B virus infection. N Engl J Med. 2008;359(14):1486–500.
4 Wedemeyer H, Manns Epidemiology MP. pathogenesis and management of hepatitis D: update and challenges ahead. Nature Rev Gastroenterol Hepatol 2010;7(1):31–40.
5 Krawitt EL. Autoimmune hepatitis. N Engl J Med. 2006;354(1):54–66.
6 Kaplan MM, Gershwin ME. Primary biliary cirrhosis. N Engl J Med. 2005;353(12):1261–73.
7 Lucey MR, Mathurin P, Morgan TR. Alcoholic hepatitis. N Engl J Med. 2009;360(26):2758–69.
8 Tujios, S. and R.J. Fontana Mechanisms of drug-induced liver injury: from bedside to bench. Nature reviews. Gastroenterology & hepatology. 2011;
9 Riehle KJ, Dan YY, Campbell JS, Fausto N. New concepts in liver regeneration. J Gastroenterol Hepatol. 2011;26(Suppl 1):203–212.
10 Friedmann S, Dantes A, Amsterdam A. Ovarian transcriptomes as a tool for a global approach of genes modulated by gonadotropic hormones in human ovarian granulosa cells. Endocrine. 2005;26(3):259–65.
11 Yeoman AD, Al-Chalabi T, Karani JB, et al. Evaluation of risk factors in the development of hepatocellular carcinoma in autoimmune hepatitis: Implications for follow-up and screening. Hepatology. 2008;48(3):863–70.
12 Cavazza A, Caballería L, Floreani A, et al. Incidence, risk factors, and survival of hepatocellular carcinoma in primary biliary cirrhosis: comparative analysis from two centers. Hepatology. 2009;50(4):1162–1168.
13 Virchow R. An Address on the Value of Pathological Experiments. Br Med J. 1881;2(1075):198–203.
14 Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357(9255):539–45.
15 Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70.
16 Demaria S, Pikarsky E, Karin M, et al. Cancer and inflammation: promise for biologic therapy. J Immunother. 2010;33(4):335–51.
17 Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454(7203):436–44.
18 Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. 2009;30(7):1073–1081.
19 Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674.
20 Llovet JM, Beaugrand M. Hepatocellular carcinoma: present status and future prospects. J Hepatol. 2003;38(Suppl 1):136–149.
21 Cover TL, Blaser MJ. Helicobacter pylori in health and disease. Gastroenterology. 2009;136(6):1863–1873.
22 Mostafa MH, Sheweita SA, O’Connor PJ. Relationship between schistosomiasis and bladder cancer. Clin Microbiol Rev. 1999;12(1):97–111.
23 Nishiyama R, Kanai T, Abe J, et al. Hepatocellular carcinoma associated with autoimmune hepatitis. J Hepatobiliary Pancreat Surg. 2004;11(3):215–9.
24 Pohl C, Hombach A, Kruis W. Chronic inflammatory bowel disease and cancer. Hepatogastroenterology. 2000;47(31):57–70.
25 Maggs JR, Chapman RW. An update on primary sclerosing cholangitis. Curr Opin Gastroenterol. 2008;24(3):377–83.
26 Fan JG, Farrell GC. Epidemiology of non-alcoholic fatty liver disease in China. J Hepatol. 2009;50(1):204–10.
27 Park EJ, Lee JH, Yu GY, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010;140(2):197–208.
28 Legakis I, Syrigos K. Obesity modulation - the role in carcinogenesis. Anticancer Agents Med Chem. 2010;10(6):481–490.
29 Wolf MJ, Seleznik GM, Zeller N, Heikenwalder M. The unexpected role of lymphotoxin beta receptor signaling in carcinogenesis: from lymphoid tissue formation to liver and prostate cancer development. Oncogene. 2010;29(36):5006–18.
30 Lukashev M, LePage D, Wilson C, et al. Targeting the lymphotoxin-beta receptor with agonist antibodies as a potential cancer therapy. Cancer Res. 2006;66(19):9617–24.
31 Vucur M, Roderburg C, Bettermann K, et al. Mouse models of hepatocarcinogenesis: what can we learn for the prevention of human hepatocellular carcinoma? Oncotarget. 2010;1(5):373–8.
32 Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140(6):883–99.
33 Coley WB. The treatment of malignant tumors by repeated inoculations of erysipelas. With a report of ten original cases. 1893. Clin Orthop Relat Res. 1991;(262):3–11.
34 Sylvester RJ. Bacillus Calmette-Guerin treatment of non-muscle invasive bladder cancer. International journal of urology: official journal of the Japanese Urological Association. 2011; 18(2):113–20.
35 Wolf MJ, Seleznik GM, Heikenwalder M. Lymphotoxin’s link to carcinogenesis: friend or foe? from lymphoid neogenesis to hepatocellular carcinoma and prostate cancer. Adv Exp Med Biol. 2011;691:231–249.
36 El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132(7):2557–2576.
37 Sherman M. Epidemiology of hepatocellular carcinoma. Oncology. 2010;78(Suppl 1):7–10.
38 Dutkowski P, et al. Current and future trends in liver transplantation in Europe. Gastroenterology. 2010; 138(3):802–9 e1–4.
39 Llovet JM, Ricci S, Mazzaferro V, et al; SHARP Investigators Study Group. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–90.
40 Cheng AL, Kang YK, Chen Z, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009;10(1):25–34.
41 Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003;362(9399):1907–7.
42 De Flora S, Bonanni P. The prevention of infection-associated cancers. Carcinogenesis. 2011
43 Luedde T, Schwabe RF. NF-kappaB in the liver – linking injury, fibrosis and hepatocellular carcinoma. Nature Rev Gastroenterol Hepatol. 2011;8(2):108–118.
44 Nakamoto Y, Guidotti LG, Kuhlen CV, Fowler P, Chisari FV. Immune pathogenesis of hepatocellular carcinoma. J Exp Med. 1998;188(2):341–350.
45 Lim IK. Spectrum of molecular changes during hepatocarcinogenesis induced by DEN and other chemicals in Fischer 344 male rats. Mech Ageing Dev. 2002;123(12):1665–1680.
46 Naugler WE, Sakurai T, Kim S, et al. Gender disparity in liver cancer due to sex differences in MyD88–dependent IL-6 production. Science. 2007;317(5834):121–124.
47 Autrup H, Wakhisi J. Detection of exposure to aflatoxin in an African population. IARC Sci Publ. 1988;(89):63–66.
48 Wang Q., Z.Y. Lin, and X.L. Feng Alterations in metastatic properties of hepatocellular carcinoma cell following H-ras oncogene transfection. World journal of gastroenterology: WJG. 2001;7(3):335–9.
49 Pikarsky E, Porat RM, Stein I, et al. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431(7007):461–466.
50 Vainer GW, Pikarsky E, Ben-Neriah Y. Contradictory functions of NF-kappaB in liver physiology and cancer. Cancer Lett. 2008;267(2):182–188.
51 Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121(7):977–990.
52 Browning JL, French LE. Visualization of lymphotoxin-beta and lymphotoxin-beta receptor expression in mouse embryos. J Immunol. 2002;168(10):5079–5087.
53 Haybaeck J, Zeller N, Wolf MJ, et al. A lymphotoxin-driven pathway to hepatocellular carcinoma. Cancer Cell. 2009;16(4):295–308.
54 Lo JC, Wang Y, Tumanov AV, et al. Lymphotoxin beta receptor-dependent control of lipid homeostasis. Science. 2007;316(5822):285–288.
55 Tumanov AV, et al. T cell-derived lymphotoxin regulates liver regeneration. Gastroenterology. 2009;136(2):694–704 e4.
56 Ruddell RG, Knight B, Tirnitz-Parker JE, et al. Lymphotoxin-beta receptor signaling regulates hepatic stellate cell function and wound healing in a murine model of chronic liver injury. Hepatology. 2009;49(1):227–239.
57 Haybaeck J, Heikenwalder M, Klevenz B, et al. Aerosols transmit prions to immunocompetent and immunodeficient mice. PLoS Pathog. 2011;7(1):e1001257.
58 Luedde T, Beraza N, Kotsikoris V, et al. Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell. 2007;11(2):119–32.
59 Bettermann K, Vucur M, Haybaeck J, et al. TAK1 suppresses a NEMO-dependent but NF-kappaB-independent pathway to liver cancer. Cancer Cell. 2010;17(5):481–496.
60 Vick B, Weber A, Urbanik T, et al. Knockout of myeloid cell leukemia-1 induces liver damage and increases apoptosis susceptibility of murine hepatocytes. Hepatology. 2009;49(2):627–36.
61 Weber A, Boger R, Vick B, et al. Hepatocyte-specific deletion of the antiapoptotic protein myeloid cell leukemia-1 triggers proliferation and hepatocarcinogenesis in mice. Hepatology. 2010;51(4):1226–36.
62 Weng SY, Yang CY, Li CC, et al. Synergism between p53 and Mcl-1 in protecting from hepatic injury, fibrosis and cancer. J Hepatol. 2011;54(4):685–694.
63 Han J, Goldstein LA, Gastman BR, Rabinowich H. Interrelated roles for Mcl-1 and BIM in regulation of TRAIL-mediated mitochondrial apoptosis. J Biol Chem. 2006;281(15):10153–63.
64 Jost PJ, Kaufmann T. Cancer caused by too much apoptosis – an intriguing contradiction? Hepatology. 2010;51(4):1110–112.
65 Zhou D, Conrad C, Xia F, et al. Mst1 and Mst2 maintain hepato-cyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell. 2009;16(5):425–438.
66 Wu S, Huang J, Dong J, Pan D. hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell. 2003;114(4):445–56.
67 Udan RS, Kango-Singh M, Nolo R, Tao C, Halder G. Hippo promotes proliferation arrest and apoptosis in the Salvador/Warts pathway. Nat Cell Biol. 2003;5(10):914–20.
68 Möröy T, Marchio A, Etiemble J, Trépo C, Tiollais P, Buendia MA. Rearrangement and enhanced expression of c-myc in hepatocellular carcinoma of hepatitis virus infected woodchucks. Nature. 1986;324(6094):276–9.
69 Santoni-Rugiu E, Preisegger KH, Kiss A, et al. Inhibition of neoplastic development in the liver by hepatocyte growth factor in a transgenic mouse model. Proc Natl Acad Sci U S A. 1996;93(18):9577–82.
70 Santoni-Rugiu E, Nagy P, Jensen MR, Factor VM, Thorgeirsson SS. Evolution of neoplastic development in the liver of transgenic mice co-expressing c-myc and transforming growth factor-alpha. Am J Pathol. 1996;149(2):407–28.
71 Terradillos O, Billet O, Renard CA, et al. The hepatitis B virus X gene potentiates c-myc-induced liver oncogenesis in transgenic mice. Oncogene. 1997;14(4):395–404.
72 Sekine S, Ogawa R, Mcmanus MT, Kanai Y, Hebrok M. Dicer is required for proper liver zonation. J Pathol. 2009;219(3):365–72.
73 Thorgeirsson SS, Santoni-Rugiu E. Transgenic mouse models in carcinogenesis: interaction of c-myc with transforming growth factor alpha and hepatocyte growth factor in hepatocarcinogenesis. Br J Clin Pharmacol. 1996;42(1):43–52.
74 Murakami H, Sanderson ND, Nagy P, Marino PA, Merlino G, Thorgeirsson SS. Transgenic mouse model for synergistic effects of nuclear oncogenes and growth factors in tumorigenesis: interaction of c-myc and transforming growth factor alpha in hepatic oncogenesis. Cancer Res. 1993;53(8):1719–23.
75 Jhappan C, Stahle C, Harkins RN, Fausto N, Smith GH, Merlino GT. TGF alpha overexpression in transgenic mice induces liver neoplasia and abnormal development of the mammary gland and pancreas. Cell. 1990;61(6):1137–46.
76 Lee GH, Merlino G, Fausto N. Development of liver tumors in transforming growth factor alpha transgenic mice. Cancer Res. 1992;52(19):5162–70.
77 Lakhtakia R, Kumar V, Reddi H, Mathur M, Dattagupta S, Panda SK. Hepatocellular carcinoma in a hepatitis B “x” transgenic mouse model: A sequential pathological evaluation. J Gastroenterol Hepatol. 2003;18(1):80–91.
78 Chisari FV, Klopchin K, Moriyama T, et al. Molecular pathogenesis of hepatocellular carcinoma in hepatitis B virus transgenic mice. Cell. 1989;59(6):1145–56.
79 Kim CM, Koike K, Saito I, Miyamura T, Jay G. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature. 1991;351(6324):317–320.
80 Moriya K, Fujie H, Shintani Y, et al. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat Med. 1998;4(9):1065–1067.
81 Sepulveda AR, Finegold MJ, Smith B, et al. Development of a transgenic mouse system for the analysis of stages in liver carcinogenesis using tissue-specific expression of SV40 large T-antigen controlled by regulatory elements of the human alpha-1–antitrypsin gene. Cancer Res. 1989;49(21):6108–17.
82 DuBois, G.C., E. Appella, and L.W. Law Isolation of a tumor-associated transplantation antigen (TATA) from an SV40–induced sarcoma. Resemblance to the TATA of chemically induced neoplasms. International journal of cancer. Journal international du cancer. 1984;34(4):561–6.
83 Mauad TH, van Nieuwkerk CM, Dingemans KP, et al. Mice with homozygous disruption of the mdr2 P-glycoprotein gene. A novel animal model for studies of nonsuppurative inflammatory cholangitis and hepatocarcinogenesis. Am J Pathol. 1994;145(5):1237–45.
84 Ghebranious N, Sell S. Hepatitis B injury, male gender, aflatoxin, and p53 expression each contribute to hepatocarcinogenesis in transgenic mice. Hepatology. 1998;27(2):383–391.
85 McGlynn KA, Hunter K, LeVoyer T, et al. Susceptibility to aflatoxin B1 – related primary hepatocellular carcinoma in mice and humans. Cancer Res. 2003;63(15):4594–601.
86 Tarsetti F, Lenzi R, Salvi R, et al. Liver carcinogenesis associated with feeding of ethionine in a choline-free diet: evidence against a role of oval cells in the emergence of hepatocellular carcinoma. Hepatology. 1993;18(3):596–603.
87 Sun FX, et al. Establishment of a metastatic model of human hepatocellular carcinoma in nude mice via orthotopic implantation of histologically intact tissues. International journal of cancer. Journal international du cancer. 1996;66(2):239–43.