Autophagy of pathogens alarms the immune system and participates in its effector functions
DOI: https://doi.org/10.4414/smw.2011.13198
Summary
Macroautophagy is a main catabolic pathway of eukaryotic cells, delivering cytoplasmic constituents for lysosomal degradation. Originally described as a starvation response, it has now been realised that macroautophagy supports many aspects of innate and adaptive immunity by facilitating innate pathogen detection and antigen presentation, as well as pathogen clearance and lymphocyte expansion. In the first half of this review, we summarise new insights into substrate selection and macroautophagic support of vesicular transport pathways, which underlie macroautophagic regulation of afferent and efferent immunity to pathogens, as outlined in the second half of the review. Applying this increased mechanistic understanding to infectious disease settings should allow us to identify further pathways for pathogen restriction, which can be explored for therapeutic manipulations of macro-autophagy.
Introduction
Eukaryotic cells primarily use two catabolic systems to degrade their cytoplasmic constituents, the proteasome and autophagy. While proteasomal degradation requires the substrates to be unfolded by chaperones, autophagy can turn over protein aggregates and even whole cell organelles in lysosomes. The later ability enables autophagy to also target cytosolic pathogens, whole bacteria and viruses [1, 2] for degradation, as an effector mechanism of adaptive and innate immunity (efferent arm of the immune response), and to deliver pathogen fragments for pathogen associated molecular pattern (PAMP) sensing and antigen presentation (afferent arm of the immune response) [3].
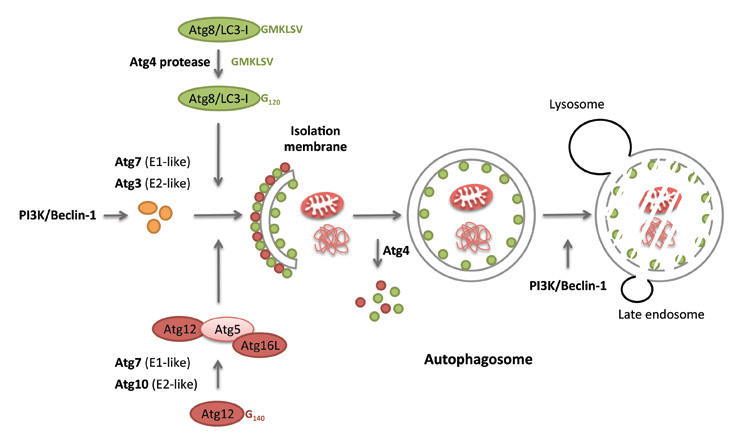
Figure 1
The two ubiquitin-like conjugation systems of macroautophagy. Autophagosomes form at PI3P marked membrane domains. At these pre-autophagosomal structures, Atg12 gets coupled to Atg5 and associates with Atg16L1 to conjugate Atg8/LC3 (after cleavage by Atg4 and activation by Atg3 and 7) to the autophagosomal membrane. Following autophagosome completion, the outer autophagosome membrane looses its Atg decoration in a Atg4-dependent manner, while Atg8/LC3, coupled to the inner autophagosomal membrane, is degraded with the autophagosome cargo after fusion with lysosomes.
Both immunological and metabolic functions of autophagy use the same molecular core machinery. Of the three autophagic pathways, macro-, micro- and chaperone-mediated autophagy, macroautophagy is best understood in this respect (fig. 1). More than 30 essential autophagy-related gene (Atg) products contribute to autophagosome formation and degradation [4, 5]. Of these, we only want to highlight three complexes, which are primarily utilised for monitoring and manipulation of macroautophagy, both by scientists and pathogens. The first of these seems to determine the site of autophagosome generation as well as catalysing autophagosome degradation and involves Atg6/Beclin-1. This protein associates with the class III phosphatidylinositol-3–OH (PI3) kinase Vps34 and the myristoylated membrane anchoring protein Vps15. Depending on the other participants in the complex, the Atg6/Vps34 can initiate autophagosome formation (together with Atg14) at the rough endoplasmic reticulum, the Golgi apparatus, mitochondria, outer nuclear or cell membrane [6–13], and augment or inhibit autophagosome fusion with lysosomes (together with UVRAG or Rubicon, respectively). Due to the crucial function of these class III PI3 kinase complexes, Atg6/Beclin-1 is modulated by many pathogens, by the anti-apoptotic proteins Bcl-2 and Bcl-XL, as well as by mutations in cancer [14–16]. The second complex, downstream of PI3 labelling of the membrane sites for autophagosome generation, is generated by the ubiquitin-like coupling of Atg12 to Atg5. This is assisted by the E1-and E2-like enzymes, Atg7 and Atg10. The Atg12 conjugate with Atg5 then associate with Atg16L1 to form an E3–like ligase for the conjugation of the ubiquitin-like molecule Atg8/LC3 to the autophagosomal membrane, after Atg8/LC3 has been proteolytically processed by Atg4 and activated by the E1- and E2-like enzymes Atg7 and Atg3. While the Atg12 complex and Atg8/LC3 get recycled from the outer autophagosomal membrane, the later by Atg4, Atg8/LC3 stays associated with the inner autophagosomal membrane and gets degraded in lysosomes together with the macroautophagy substrates. Atg8/LC3 supports membrane hemifusion during the elongation of the autophagosomal membrane and forms the membrane anchor for substrate recruitment into the autophagosomal membrane [17, 18]. This core machinery allows flexible formation of small autophagosomes (0.5–1 μm in diameter) for metabolic purposes, as well as large xenophagosomes (up to 10 μm in diameter) for the engulfment of cytosolic bacteria, using membranes from multiple sources.
Substrate selection for autophagy
While macroautophagy was originally considered a non-discriminatory degradation process for cytoplasmic material in lysosomes, it has recently become apparent that its different substrates are selectively imported into autophagosomes and that, even during starvation, a hierarchy of sequential cytoplasm degradation exists [19]. Interestingly, poly-ubiquitination, which also targets soluble substrates to proteasomal degradation, selects substrates for macroautophagy towards lysosomal degradation. This selection of poly-ubiquitinated substrates into autophagosomes seems to be mediated by proteins that contain both ubiquitin binding domains (UBA or UBZ) and LC3 interacting motifs (LIRs: WXXL or WXXI), and directly anchor the macroautophagy substrates to the inner autophagosomal membrane. Three such proteins have so far been identified in mammalian cells: p62/sequestosome 1, NBR1 and NDP52 [18, 20–22]. They mediate the recruitment of protein aggregates, modified by the chaperone- associated ubiquitin ligase CHIP, into autophagosomes. The chaperone BAG3 cooperates with p62/sequestosome 1 in the macroautophagy of these poly-ubiquitinated protein aggregates [23–26]. Furthermore, p62/sequestosome 1, assisted by its interacting protein Alfy, promotes protein aggregation for macroautophagic substrate generation [27, 28]. In addition to protein aggregates, ubiquitination of mitochondria via the ubiquitin ligase Parkin might facilitate their recruitment to autophagosomes [29]. In contrast to higher eukaryotes, the yeast Saccharomyces cerevisiae has no obvious homologues of p62/sequestosome 1, NBR1 and NDP52 [30], and ribosomal degradation by macroautophagy is even enhanced by deubiquitination [31]. Instead, the yeast expresses LIR containing proteins like Atg9 that directly interact with enzymes, such as aminopeptidase 1 and α-mannosidase 1, which are imported into the lyosomal vacuole by the macroautophagy-related biosynthetic cytoplasm-to-vacuole (Cvt) pathway [32]. Furthermore, in yeast, Atg32 was reported to recruit mitochondria to autophagosomes [33, 34]. This later mechanism seems to be conserved, since the outer mitochondrial membrane protein NIX of mammalian cells also contains a LIR and assists in macroautophagy of mitochondria [35, 36]. Thus, both poly-ubiquitination dependent and independent substrate recruitment to macroautophagy exist. While yeast seems to employ LIR containing proteins that directly bind to autophagosome cargo, mammalian cells have, in addition, developed proteins that cross-link ubiquitin and LC3 to allow macroautophagy of poly-ubiquitinated substrates. This allows a much more versatile use of macroautophagy, utilising the sophisticated mammalian system of ubiquitin ligases for protein degradation.
Vesicular transport pathways assisted by autophagy
The eukaryotic endomembrane system includes endoplasmic reticulum (ER), Golgi and lysosomes. Macromolecules, such as proteins and lipids, are exchanged between these organelles and the extracellular space through vesicular transport. This movement is bidirectional and therefore vesicles can deliver their cargo from trans-Golgi to storage vesicles, to the lysosomes or to the plasma membrane, or follow the reverse route [37]. Growing evidence, which is summarised below, demonstrates that autophagy can assist both secretory and endocytic pathways. This may not be so surprising considering that the machinery required for macroautophagy, especially for autophagosome maturation, overlaps with other membrane trafficking pathways. This is the case for some subunits of the endosomal sorting complex required for transport (ESCRT), like for example Vps4, Vps25, Vps28 or Vps32, which have been shown to be essential for generation of fully degradative autophagosomes in flies [38]. Furthermore, Vps34, a PI3K that was first described to target proteins containing the phosphoinositide-binding domain to endosomal/lysosomal vesicles, regulates both autophagosome generation and maturation in yeast as well as in mammalian cells [39]. The cross-talk between classical vesicular transport and autophagy is further exemplified by the Rab family of proteins. Rabs are peripheral membrane-bound GTPases that regulate membrane trafficking (vesicle formation, movement and fusion). A recent study using fly models suggests that Rab5 supports early macroautophagy processes in a way that is independent of its endocytic functions [40]. Pre-autophagosomal structures seem to be assembled around complexes containing Rab5, Vps34 and Beclin-1. Since inhibition of Rab5 decreases autophagosome formation, but leads to accumulation of autophagosome precursors, the authors suggested that Rab5 would act as an activator of Vps34, an essential component for the progression of autophagosome formation. Moreover, late endosomal Rabs, such as Rab7 and Rab11, were proposed to promote autophagosome fusion with lysosomes and multi-vesicular bodies (MVB), respectively [41, 42], further supporting the intersection of both vesicular trafficking and macroautophagy machinery. Thus, macroautophagy seems to make use of common mechanisms and components of vesicular trafficking.
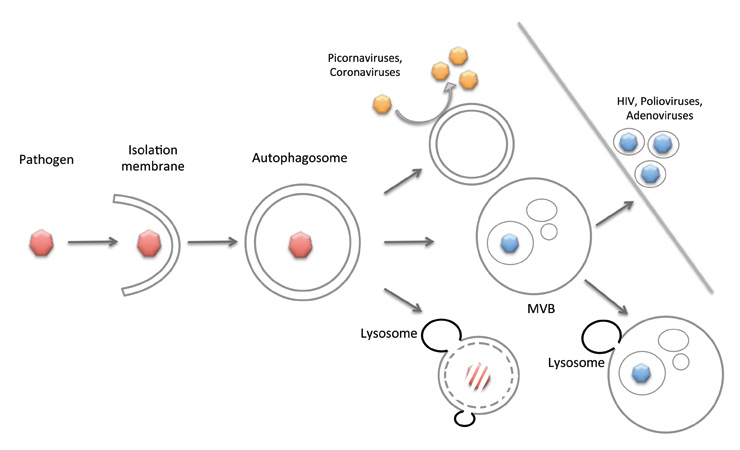
Figure 2
Macroautophagy in pathogen restriction and replication. Pathogens, like viruses, bacteria and parasites, can be degraded by macroautophagy, or might use autophagosomal delivery to multi-vesicular bodies for their release from the host cell. In addition, viruses re-model cytoplasmic membranes via macroautophagy in order to replicate on their surface.
Apart from the shared use of this molecular machinery by macroautophagy and other vesicular transport processes, there are recent data suggesting that proteins essential for macroautophagy can effectively assist exocytosis. This is the case for the unconventional secretion of acyl co-enzyme A-binding protein (Acb1) in Saccharomyces cerevisae,which requires Atg5, 7, 8 and 12, but not secretory pathway components such as Sec1, 7 and 23 [43]. This study also suggested that autophagosomes sequester Acb1 but do not proceed to normal maturation by fusing with the lysosomal compartment (yeast vacuole). Instead, Acb1–containg autophagosomes are redirected to the plasma membrane, in a process involving MVBs and the t-SNARE Sso1, ultimately releasing their content in the extracellular space. This interpretation is supported by the fact that deletion of essential mediators of autophagosomal fusion with the vacuolar membrane (Vam3 and Ypt7) does not inhibit Acb1 secretion. Another study making use of yeast genetics further supports a role for macroautophagy in unconventional protein secretion. Indeed, Manjithaya et al. showed that, similar to what was observed in S. cerevisae,in the yeast Pichia pastoris core Atg proteins (Atg1, 6, 8 and 17) are necessary for Acb1 secretion [44]. Moreover, adding rapamycin to cell cultures enhanced Acb1 secretion in wild type cells but not in Atg1 knock-out mutants. Together, these studies provide evidence for a role of macroautophagy in unconventional protein secretion. However, direct proof of the involvement of whole autophagosomes capturing Acb1 as a specific substrate has yet to be obtained. Concerning macroautophagy-assisted exocytosis, some other examples are worth mentioning. For instance, macroautophagy seems to contribute to non-lytic viral release to the extracellular space (fig. 2). This has been suggested for polio viruses, adenoviruses and the human immunodeficiency virus, based on the observation that following infection double membrane vesicles, similar to autophagosomes, accumulate in the cytoplasm and even without breakdown of the host cell plasma membrane, a significant amount of virions can be found in culture medium [45–47]. Furthermore, it was recently suggested that corona-viruses induce formation of autophagosome-like vesicles that seem to be dependent on LC3–I but, intriguingly, not on its lipidated form (LC3–II) or on any other essential macroautophagy gene products [48]. So far we can only speculate that LC3–I could then act as a coating protein that would signal vesicle biogenesis. Altogether, these data suggest that macroautophagy facilitates exocytosis during unconventional protein secretion and viral replication.
In the opposite direction, macroautophagy has been reported to assist phagosomal maturation [49, 50]. This process was suggested to depend on recruitment of LC3 and Beclin-1 to phagosomes, which will then, through a still unknown mechanism, promote lysosomal fusion to the phagosome, boosting phagosomal content degradation. As with coronavirus replication, parts of the macroautophagy machinery seem to be used for phagosome maturation, and elucidation of the role of Atgs in these vesicular transport pathways will probably also define their function during classical macroautophagy more clearly.
Pathogen restriction by autophagy
In its more classical form, macroautophagy as a catabolic pathway can directly target and destroy intracellular pathogens (fig. 2). Indeed, several studies report an increased susceptibility of infection following deletion of macroautophagy components. However, initial signalling events that lead to microbial engulfment by isolation membranes remain poorly understood. Still, one may speculate that these involve poly-ubiquitination or Atg recruitment domains, as previously addressed in section 2 of this review. Examples of pathogen restriction by macroautophagy can be found in bacterial, parasitic and, to a lesser extent, in viral infections. For example, Listeria monocytogenes evades the extreme conditions of the phagosome to grow in the cytosol, by escaping through listeriolysin-O (LLO) formed pores in the phagosomal membrane. The damage induced to the phagosomal membrane was suggested as a trigger for macroautophagy [51]. In the cytosol, Listeria surrounds itself with an actin coat to escape engulfment by autophagosomes, and only ActA- deficient bacteria are efficiently degraded by macroautophagy [52]. Similarly, Shigella expresses the IcsB protein that blocks binding of Atg5 to its VirG protein to prevent incorporation into autophagosomes [53]. In contrast, however, many other cytosolic pathogens, like group A Streptococcus, Francisella tularensis or Rickettsia conorii,are restricted by macroautophagy and this innate defence is defective in Atg5- deficient macrophages [1, 5]. Apart from targeting cytosolic microbes, macroautophagy can also influence the control of pathogens, which live and replicate inside vesicular compartments. This is the case for Mycobacterium tuberculosis, macrophage phagosome-resident bacteria that are capable of interfering with phagosomal maturation pathways. Initial studies in murine cell lines demonstrated that induction of autophagy can counteract the pathogen-induced remodelling of phagosomes by promoting phagosome acidification and subsequent mycobacteria clearance [54]. This process involves the immunity-related GTPase family M (IRGM), and in accordance, human genetic approach studies identified protective IRGM gene variants against M. tuberculosis[55]. Furthermore, several other macroautophagy genes have been found in a recent M. tuberculosis resistance gene screen [56]. In addition to mycobacteria, the protozoan Toxoplasma gondii, which also conditions phagosomes as its replication niche, can either be directly targeted by autophagosomes following disruption of the vesicle harbouring the parasite, or the whole parasitophorous vacuole can be re-routed to lysosomes in an autophagy-dependent fashion [50, 57]. Thus, macroautophagy can restrict bacterial and parasitic pathogens, but some of them have developed escape mechanisms against this mechanism of innate immunity.
In addition to the strategies presented above, autophagy can also act on the reduction of bacterial load through the delivery of sources for antimicrobial peptides to microbe-containing vesicles. Some of the bactericidal components identified so far have been ubiquitin-derived peptides, which are generated by lysosomal proteolysis after delivery to bacteria containing phagosomes via macroautophagy. These are either directly derived from poly-ubiquitinated autophagosome content or Fau, a ribosomal protein containing an ubiquitin domain [58, 59]. In the case of Fau, p62/sequestosome 1 links this source of bactericidal peptides to LC3 for engulfment by autophagosomes. Thus, macroautophagy does not only restrict bacterial pathogens by degradation in lysosomes, but also increases bactericidal activity of these compartments by delivering proteins, which are processed to bactericidal peptides.
In addition to innate restriction of bacterial and parasitic infections, macroautophagy has also been reported to restrict viral infections, although the underlying mechanisms are still mostly unclear. The existing studies have mainly established a correlation between Atg gene function and control of viral replication. For example, the plant mosaic virus infection seems to be controlled by Atg3, Atg7, Beclin-1 and Vps34 [60]. Similarly, depletion of Atg5, Atg8a, Atg18 and other autophagy related genes significantly increased vesicular stomatitis viral loads in Drosophila[61]. In addition to these invertebrate in vivo systems, neuronal control of herpes simplex virus (HSV) or Sindbis virus infections by macroautophagy has been recently described [62, 63]. HSV that lacked the macroautophagy-inhibitory domain of the ICP34.5 protein, replicated to lower viral loads and caused decreased pathology after intracerebral infection. Similarly, Sindbis virus infection in the central nervous system was aggravated if macroautophagy was compromised by diminishing Atg5 levels in infected neurons. In the later study, the authors demonstrated that Atg5 silencing was not generally required for Sindbis virus replication in immortalised cell lines, however, neuronal Atg5 function was rather important in clearance of Sindbis virus antigens and for protection of neurons against cell death. Therefore, macroautophagy does not seem to directly impact pathogen replication, but rather promotes host cell resistance to infection.
Innate pathogen sensing via autophagy
In addition to its direct role in intracellular pathogen targeting, macroautophagy can also regulate the microbial fate by mediating pathogen detection and influencing the host cell inflammatory responses. This was proposed for the recognition of vesicular stomatitis virus (VSV) by toll-like receptor 7 (TLR7) in plasmacytoid dendritic cells (pDCs), which is negatively influenced upon inhibition of macroautophagy [64]. Indeed, after VSV infection, both pharmacological inhibition of macroautophagy and Atg5 knock-out in murine cells, resulted in diminished secretion of IFN-α: a hallmark cytokine in response to RNA virus infection. Furthermore, delivery of B cell receptor ligands to TLR9 containing vesicles for CpG DNA detection was found to be dependent on macroautophagy [65]. In contrast to pDCs and B cells, other cell types sense viral genomes and transcription products through cytosolic receptors of the Rig-1–like receptors family (RLRs). Jounai et al. [66] demonstrated that in mouse embryonic fibroblasts (MEFs) the absence of Atg5 significantly increased type I IFN production in response to dsRNA. According to the authors, the Atg5–Atg12 complex suppresses IFN-α production through direct interaction with the CARD domain of both IPS-1 and RIG-I. In agreement, a more recent study demonstrated that Atg5–/– MEFs and primary murine liver macrophages produce higher type I IFN levels upon RIG-I stimulation [67]. This enhancement of RLR signalling in the absence of macroautophagy is linked to the accumulation of dysfunctional mitochondria, which present an unbalanced redox status, leading to augmented reactive oxygen species (ROS) levels that can directly stimulate RLR. Similarly, absence of macroautophagy enhances inflammasome activation [68], and this also seems to require mitochondrial ROS production [69, 70]. Thus, depending on the pathogen associated molecular pattern (PAMP) and the detecting cell, macroautophagy can either restrict or augment the PAMP recognition signalling mechanisms.
In addition to virus detection, macroautophagy also regulates bacterial sensing. Following Shigella infection, phagosome rupture generates membrane remnants that are targeted for elimination by macroautophagy due to a poly-ubiquitination process that recruits LC3 and p62 [71]. With this, macroautophagy can silence the signalling cascades activated by PAMPs and danger-associated molecular patterns (DAMPs) contained in the Shigella vacuole membrane remnants, including NF-κB-dependent cytokine production, ROS production and cell death. Therefore, by eliminating signal molecules implicated in adverse host cell effects, macroautophagy might work as a pro-survival factor following bacterial infections.
On the other hand, innate immune sensing pathways can induce the macroautophagy pathway. Along these lines, stimulation of cells with several TLR agonists, such as Pam3Cys4, zymosan, polyinosinic:polycytidylic acid, lipopolysaccharide, single stranded RNA and imiquimod, was suggested to up-regulate macroautophagy [5]. Since most of these studies were performed in the murine Raw264.7 cell line, one must be cautious when generalising these findings to other cell types, especially to primary cells. In fact, there are some conflicting data regarding the TLR-dependent induction of macroautophagy. Immunofluorescence analysis of primary macrophages from C57BL/6 mice treated with TLR2, 3, 4, 5, 7, 8 and 9 agonists did not induce an increase in LC3 positive vesicles [68]. However, this contrasts with what was observed in primary murine macrophages of Atg7+/–mice [49], in bone-marrow-derived macrophages from knock-in GFP-LC3 transgenic mice [72], and in primary human alveolar macrophages [73]. Especially in the later cell population, TLR4 and TLR7 stimulation have been suggested to target vacuolar mycobacteria for degradation [72, 73]. Apart from TLRs, stimulation of other pattern recognition receptors also regulates macroautophagy. In DCs, NOD2 receptor stimulation with muramyldipeptide induces macroautophagy in an Atg5-, 7- and 16L1-dependent manner [74]. Here, NOD2–induced macroautophagy results in bacteria restriction and increased MHC class II antigen presentation. Interestingly, both NOD2 and Atg16L1 were identified as susceptibility genes for Crohn’s disease [75–78], and patients’ DCs are deficient in MHC class II up-regulation after NOD2 stimulation [74]. In conclusion, these studies suggest that specific PAMP recognition can regulate macroautophagy.
Support of adaptive immunity by autophagy
The degradation in lysosomes via macroautophagy also generates proteolytic fragments that can be presented on MHC molecules to T cells (fig. 3). In this respect, CD4+, so-called helper T cells (the orchestrators of adaptive immunity), primarily recognise lysosomal degradation products presented on MHC class II molecules, while CD8+, so-called cytotoxic T cells (the main effectors of cell-mediated adaptive immune responses), are stimulated by mainly proteasomal products presented on MHC class I molecules. Accordingly, macroautophagy has primarily been documented to deliver antigens for MHC class II presentation [79]. This includes cytosolic model antigens [80–82], viral [83–85], intracellular bacteria derived [86, 87] and self-antigens [88]. The self-antigen presentation on MHC class II molecules after macroautophagy by thymic epithelial cells has been shown to shape both positive and negative T cell selection in mice [89]. In the absence of Atg5, some clonal CD4+, but not CD8+, T cell specificities were inefficiently positively selected on cortical thymic epithelial cells, and a T cell repertoire was generated that was inefficiently tolerised by negative selection on medullary thymic epithelial cells, and, therefore, elicited autoimmune pathology in several tissues. In addition to this role for macroautophagy in self-antigen presentation on MHC class II molecules during T cell education, macroautophagy enhanced mycobacterial antigen presentation in DCs for vaccination [86], and was also essential in DCs to prime strong CD4+ T cell responses during HSV infection [90]. During HSV infection, the viral gene product ICP34.5 antagonises macroautophagy, and ICP34.5 deficient HSV infection primes elevated CD4+ T cell responses, which control viral titers at lower levels [91]. These studies suggest that macroautophagy contributes to adaptive immune responses by facilitating pathogen-derived antigen processing for MHC class II presentation, and shaping the CD4+ T cell repertoire via self-antigen presentation on MHC class II molecules in the thymus.
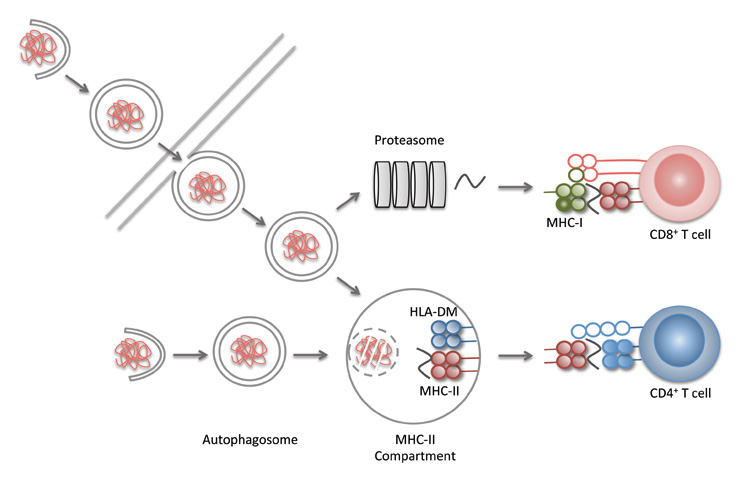
Figure 3
Macroautophagy in antigen processing for MHC presentation. Proteasomal products are primarily presented on MHC class I molecules to CD8+ T cells, while lyosomal products are preferentially presented on MHC class II molecules to CD4+ T cells. Macroautophagy seems to contribute to antigen presentation in at least three ways: i) it delivers cytoplasmic antigens for MHC class II presentation, ii) enhances phagosome maturation and iii) packages antigens in bystander cells for more efficient cross-presentation by antigen presenting cells.
Apart from the fairly extensive evidence for a role of macroautophagy in MHC class II antigen presentation, a few reports also suggest that this pathway can assist in MHC class I presentation of antigens. It was suggested that macroautophagy is required, late during HSV infection, for intracellular antigen processing onto MHC class I molecules [10]. Surprisingly, the authors proposed that autophagosome contained antigens would leave these vesicles again for further proteasomal processing and loading onto MHC class I molecules for CD8+ T cell stimulation. Furthermore, cross-presentation of antigens for MHC class I presentation by bystander antigen presenting cells was proposed to require macroautophagy in the antigen donor cell [92, 93]. The mechanism of this antigen packaging for efficient cross-presentation, however, remains unclear. Thus, macroautophagy might, under certain conditions, also contribute to antigen processing for MHC class I presentation to CD8+ T cells.
In addition to this role in antigen presentation, macroautophagy also seems to be required for survival of lymphocyte populations during development and immune responses. T cells particularly require macroautophagy for surviving thymic education and expansion during immune challenges [94]. Atg7-deficient T cells and their precursors accumulate mitochondria and expanded endoplasmic reticulum [95, 96]. The accumulation of damaged mitochondria elevates ROS levels, resulting in decreased survival of developing and stimulated macroautophagy-deficient T cells. In addition to T cells, B cells, in particular B-1a cells, require macroautophagy for their development [97]. In contrast to the macroautophagy dependency of haematopoietic progenitor cell maintenance and lymphocyte development [98–100], macrophages, conventional DCs and pDCs do seem to develop normally in the absence of macroautophagy [64, 90, 101]. This offers the possibility to investigate adaptive immune responses for roles of macroautophagy in antigen processing and pathogen sensing by myeloid cells, without compromised reactivity of macroautophagy-deficient lymphocytes.
Conclusions
Macroautophagy interfaces with many aspects of innate and adaptive immunity (fig. 4), both for pathogen sensing and elimination. While in vivoimmunological restriction of these bacteria, viruses and parasites is, mechanistically, still poorly understood, the new insights into substrate selection by macroautophagy and its support for various vesicular trafficking pathways now allow researchers to dissect which pathogens are especially prone to be targeted by macroautophagy and which cellular pathways are used. This should permit us to identify infectious disease settings, in which macroautophagy manipulation can be explored for clinical benefits.
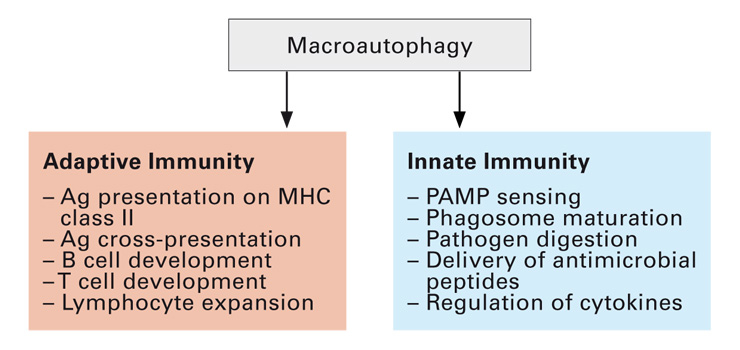
Figure 4
Macroautophagy contributes to both adaptive and innate immunity. Antigen presentation and lymphocyte populations are regulated by macroautophagy during adaptive immune responses. In addition, pathogen sensing and their degradation are also supported by this catabolic pathway.
References
1 Nakagawa I, Amano A, Mizushima N, Yamamoto A, Yamaguchi H, et al. Autophagy defends cells against invading group A Streptococcus. Science. 2004;306(5698):1037–40.
2 Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol. 2007;7(10):767–77.
3 Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature 2011;469(7330):323–35.
4 Klionsky DJ, Codogno P, Cuervo AM, Deretic V, Elazar Z, Fueyo-Margareto J, Gewirtz DA, Kroemer G, Levine B, Mizushima N, Rubinsztein DC, Thumm M, Tooze SA. A comprehensive glossary of autophagy-related molecules and processes. Autophagy. 2010;6(4).
5 Münz C. Enhancing immunity through autophagy. Annu Rev Immunol. 2009;27:423–9.
6 Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol. 2009 Dec;11(12):1433–7.
7 Yla-Anttila P, Vihinen H, Jokitalo E, Eskelinen EL. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy. 2009;5(8):1180–5.
8 Yen WL, Shintani T, Nair U, Cao Y, Richardson BC, et al. The conserved oligomeric Golgi complex is involved in double-membrane vesicle formation during autophagy. J Cell Biol. 2010;188(1):101–14.
9 Lynch-Day MA, Bhandari D, Menon S, Huang J, Cai H, Bartholomew CR, Brumell JH, Ferro-Novick S, Klionsky DJ. Trs85 directs a Ypt1 GEF, TRAPPIII, to the phagophore to promote autophagy. Proc Natl Acad Sci U S A. 2010;107(17):7811–6.
10 English L, Chemali M, Duron J, Rondeau C, Laplante A, et al. Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat Immunol. 2009;10(5):480–7.
11 Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, et al. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. 2010;141(4):656–67.
12 He C, Song H, Yorimitsu T, Monastyrska I, Yen WL, Legakis JE, Klionsky DJ. Recruitment of Atg9 to the preautophagosomal structure by Atg11 is essential for selective autophagy in budding yeast. J Cell Biol. 2006;175(6):925–35.
13 Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol. 2010;12(8):747–57.
14 Gannage M, Rämer PC, Münz C. Targeting Beclin 1 for viral subversion of macroautophagy. Autophagy. 2010;6(1):166–7.
15 Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1–dependent autophagy. Cell. 2005;122(6):927–39.
16 Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003;100(25):15077–82.
17 Nakatogawa H, Ichimura Y, Ohsumi Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell. 2007;130(1):165–78.
18 Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171(4):603–14.
19 Kristensen AR, Schandorff S, Hoyer-Hansen M, Nielsen MO, Jaattela M, et al. Ordered organelle degradation during starvation-induced autophagy. Mol Cell Proteomics. 2008;7(12):2419–28.
20 Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282(33):24131–45.
21 Kirkin V, Lamark T, Sou YS, Bjorkoy G, Nunn JL, Bruun JA, Shvets E, McEwan DG, Clausen TH, Wild P, Bilusic I, Theurillat JP, Overvatn A, Ishii T, Elazar Z, Komatsu M, et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell. 2009;33(4):505–16.
22 Thurston TL, Ryzhakov G, Bloor S, von Muhlinen N, Randow F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol. 2009;10(11):1215–21.
23 Gamerdinger M, Hajieva P, Kaya AM, Wolfrum U, Hartl FU, Behl C. Protein quality control during aging involves recruitment of the macroautophagy pathway by BAG3. Embo J. 2009;28(7):889–901.
24 Arndt V, Dick N, Tawo R, Dreiseidler M, Wenzel D, et al. Chaperone-assisted selective autophagy is essential for muscle maintenance. Curr Biol. 2010;20(2):143–8.
25 Carra S, Seguin SJ, Lambert H, Landry J. HspB8 chaperone activity toward poly(Q)-containing proteins depends on its association with Bag3, a stimulator of macroautophagy. J Biol Chem. 2008;283(3):1437–44.
26 Kettern N, Rogon C, Limmer A, Schild H, Hohfeld J. The Hsc/Hsp70 Co-Chaperone Network Controls Antigen Aggregation and Presentation during Maturation of Professional Antigen Presenting Cells. PLoS ONE. 2011;6(1):e16398.
27 Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131(6):1149–63.
28 Simonsen A, Birkeland HC, Gillooly DJ, Mizushima N, Kuma A, et al. Alfy, a novel FYVE-domain-containing protein associated with protein granules and autophagic membranes. J Cell Sci. 2004;117(Pt 18):4239–51.
29 Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8(1):e1000298.
30 Kraft C, Peter M, Hofmann K. Selective autophagy: ubiquitin-mediated recognition and beyond. Nat Cell Biol. 2010;12(9):836–41.
31 Kraft C, Deplazes A, Sohrmann M, Peter M. Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nat Cell Biol. 2008;10(5):602–10.
32 He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93.
33 Okamoto K, Kondo-Okamoto N, Ohsumi Y. Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev Cell. 2009;17(1):87–97.
34 Kanki T, Wang K, Cao Y, Baba M, Klionsky DJ. Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev Cell. 2009;17(1):98–109.
35 Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, Wang J. Essential role for Nix in autophagic maturation of erythroid cells. Nature. 2008 May 4.
36 Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, Kundu M, Opferman JT, Cleveland JL, Miller JL, Ney PA. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci U S A. 2007;104(49):19500–5.
37 Anitei M, Wassmer T, Stange C, Hoflack B. Bidirectional transport between the trans-Golgi network and the endosomal system. Mol Membr Biol. 2010;27(8):443–56.
38 Longatti A, Orsi A, Tooze SA. Autophagosome formation: not necessarily an inside job. Cell Res. 2010;20(11):1181–4.
39 Backer JM. The regulation and function of Class III PI3Ks: novel roles for Vps34. Biochem J. 2008;410(1):1–17.
40 Ravikumar B, Imarisio S, Sarkar S, O’Kane CJ, Rubinsztein DC. Rab5 modulates aggregation and toxicity of mutant huntingtin through macroautophagy in cell and fly models of Huntington disease. J Cell Sci. 2008;121(Pt 10):1649–60.
41 Fader CM, Sanchez D, Furlan M, Colombo MI. Induction of autophagy promotes fusion of multivesicular bodies with autophagic vacuoles in K562 cells. Traffic. 2008;9(2):230–50.
42 Jager S, Bucci C, Tanida I, Ueno T, Kominami E, Saftig P, Eskelinen EL. Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci. 2004;117(Pt 20):4837–48.
43 Duran JM, Anjard C, Stefan C, Loomis WF, Malhotra V. Unconventional secretion of Acb1 is mediated by autophagosomes. J Cell Biol. 2010;188(4):527–36.
44 Manjithaya R, Anjard C, Loomis WF, Subramani S. Unconventional secretion of Pichia pastoris Acb1 is dependent on GRASP protein, peroxisomal functions, and autophagosome formation. J Cell Biol. 2010 Feb 22;188(4):537–46.
45 Taylor MP, Burgon TB, Kirkegaard K, Jackson WT. Role of microtubules in extracellular release of poliovirus. J Virol. 2009;83(13):6599–609.
46 Jiang H, White EJ, Gomez-Manzano C, Fueyo J. Adenovirus’s last trick: you say lysis, we say autophagy. Autophagy. 2008;4(1):118–20.
47 Kyei GB, Dinkins C, Davis AS, Roberts E, Singh SB, Dong C, Wu L, Kominami E, Ueno T, Yamamoto A, Federico M, Panganiban A, Vergne I, Deretic V. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J Cell Biol. 2009;186(2):255–68.
48 de Haan CA, Molinari M, Reggiori F. Autophagy-independent LC3 function in vesicular traffic. Autophagy. 2010;6(7):994–6.
49 Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, Komatsu M, Tanaka K, Cleveland JL, Withoff S, Green DR. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007;450(7173):1253–7.
50 Andrade RM, Wessendarp M, Gubbels MJ, Striepen B, Subauste CS. CD40 induces macrophage anti-Toxoplasma gondii activity by triggering autophagy-dependent fusion of pathogen-containing vacuoles and lysosomes. J Clin Invest. 2006;116(9):2366–77.
51 Meyer-Morse N, Robbins JR, Rae CS, Mochegova SN, Swanson MS, Zhao Z, Virgin HW, Portnoy D. Listeriolysin O is necessary and sufficient to induce autophagy during Listeria monocytogenes infection. PLoS ONE 2010;5(1):e8610.
52 Yoshikawa Y, Ogawa M, Hain T, Yoshida M, Fukumatsu M, Kim M, Mimuro H, Nakagawa I, Yanagawa T, Ishii T, Kakizuka A, Sztul E, Chakraborty T, Sasakawa C. Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nat Cell Biol. 2009;11(10):1233–40.
53 Ogawa M, Yoshimori T, Suzuki T, Sagara H, Mizushima N, Sasakawa C. Escape of Intracellular Shigella from Autophagy. Science. 2005;307(5710):727–31.
54 Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy Is a Defense Mechanism Inhibiting BCG and Mycobacterium tuberculosis Survival in Infected Macrophages. Cell. 2004;119(6):753–66.
55 Intemann CD, Thye T, Niemann S, Browne EN, Amanua Chinbuah M, Enimil A, Gyapong J, Osei I, Owusu-Dabo E, Helm S, Rusch-Gerdes S, Horstmann RD, Meyer CG. Autophagy gene variant IRGM -261T contributes to protection from tuberculosis caused by Mycobacterium tuberculosis but not by M. africanum strains. PLoS Pathog. 2009;5(9):e1000577.
56 Kumar D, Nath L, Kamal MA, Varshney A, Jain A, Singh S, Rao KV. Genome-wide analysis of the host intracellular network that regulates survival of Mycobacterium tuberculosis. Cell. 2010;140(5):731–43.
57 Ling YM, Shaw MH, Ayala C, Coppens I, Taylor GA, Ferguson DJ, Yap GS. Vacuolar and plasma membrane stripping and autophagic elimination of Toxoplasma gondii in primed effector macrophages. J Exp Med. 2006 Sep 4;203(9):2063–71.
58 Alonso S, Pethe K, Russell DG, Purdy GE. Lysosomal killing of Mycobacterium mediated by ubiquitin-derived peptides is enhanced by autophagy. Proc Natl Acad Sci U S A. 2007;104(14):6031–6.
59 Ponpuak M, Davis AS, Roberts EA, Delgado MA, Dinkins C, Zhao Z, Virgin HWt, Kyei GB, Johansen T, Vergne I, Deretic V. Delivery of cytosolic components by autophagic adaptor protein p62 endows autophagosomes with unique antimicrobial properties. Immunity. 2010 Mar 26;32(3):329–41.
60 Liu Y, Schiff M, Czymmek K, Talloczy Z, Levine B, Dinesh-Kumar SP. Autophagy regulates programmed cell death during the plant innate immune response. Cell. 2005;121(4):567–77.
61 Shelly S, Lukinova N, Bambina S, Berman A, Cherry S. Autophagy is an essential component of Drosophila immunity against vesicular stomatitis virus. Immunity. 2009;30(4):588–98.
62 Orvedahl A, Alexander D, Talloczy Z, Sun Q, Wei Y, Zhang W, Burns D, Leib D, Levine B. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host & Microbe. 2007;1:23–35.
63 Orvedahl A, MacPherson S, Sumpter R, Jr., Talloczy Z, Zou Z, Levine B. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe. 2010;7(2):115–27.
64 Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science. 2007 Mar 9;315(5817):1398–401.
65 Chaturvedi A, Dorward D, Pierce SK. The B cell receptor governs the subcellular location of Toll-like receptor 9 leading to hyperresponses to DNA-containing antigens. Immunity. 2008;28(6):799–809.
66 Jounai N, Takeshita F, Kobiyama K, Sawano A, Miyawaki A, Xin KQ, Ishii KJ, Kawai T, Akira S, Suzuki K, Okuda K. The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc Natl Acad Sci U S A. 2007;104(35):14050–5.
67 Tal MC, Sasai M, Lee HK, Yordy B, Shadel GS, Iwasaki A. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc Natl Acad Sci U S A. 2009;106(8):2770–5.
68 Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, Tanaka K, Kawai T, Tsujimura T, Takeuchi O, Yoshimori T, Akira S. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008 Oct 5.
69 Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221–5.
70 Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, Fitzgerald KA, Ryter SW, Choi AM. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12(3):222–30.
71 Dupont N, Lacas-Gervais S, Bertout J, Paz I, Freche B, Van Nhieu GT, van der Goot FG, Sansonetti PJ, Lafont F. Shigella phagocytic vacuolar membrane remnants participate in the cellular response to pathogen invasion and are regulated by autophagy. Cell Host Microbe. 2009 Aug 20;6(2):137–49.
72 Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V. Toll-like receptors control autophagy. Embo J. 2008;27(7):1110–21.
73 Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, Eissa NT. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity. 2007;27(1):135–44.
74 Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, Ferguson DJ, Campbell BJ, Jewell D, Simmons A. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med. 2010;16(1):90–7.
75 Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, Albrecht M, Mayr G, De La Vega FM, Briggs J, Gunther S, Prescott NJ, Onnie CM, Hasler R, Sipos B, Folsch UR, Lengauer T, Platzer M, Mathew CG, Krawczak M, Schreiber S. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39(2):207–11.
76 Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet 2007;39(5):596–604.
77 Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, Almer S, Tysk C, O’Morain CA, Gassull M, Binder V, Finkel Y, Cortot A, Modigliani R, Laurent-Puig P, Gower-Rousseau C, Macry J, Colombel JF, Sahbatou M, Thomas G. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411(6837):599–603.
78 Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, Britton H, Moran T, Karaliuskas R, Duerr RH, Achkar JP, Brant SR, Bayless TM, Kirschner BS, Hanauer SB, Nunez G, Cho JH. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature. 2001;411(6837):603–6.
79 Schmid D, Pypaert M, Münz C. MHC class II antigen loading compartments continuously receive input from autophagosomes. Immunity. 2007;26:79–92.
80 Brazil MI, Weiss S, Stockinger B. Excessive degradation of intracellular protein in macrophages prevents presentation in the context of major histocompatibility complex class II molecules. Eur J Immunol. 1997;27(6):1506–14.
81 Nimmerjahn F, Milosevic S, Behrends U, Jaffee EM, Pardoll DM, Bornkamm GW, Mautner J. Major histocompatibility complex class II-restricted presentation of a cytosolic antigen by autophagy. Eur J Immunol. 2003;33(5):1250–9.
82 Riedel A, Nimmerjahn F, Burdach S, Behrends U, Bornkamm GW, Mautner J. Endogenous presentation of a nuclear antigen on MHC class II by autophagy in the absence of CRM1–mediated nuclear export. Eur J Immunol. 2008;38(8):2090–5.
83 Paludan C, Schmid D, Landthaler M, Vockerodt M, Kube D, Tuschl T, Münz C. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science. 2005;307(5709):593–6.
84 Leung CS, Haigh TA, Mackay LK, Rickinson AB, Taylor GS. Nuclear location of an endogenously expressed antigen, EBNA1, restricts access to macroautophagy and the range of CD4 epitope display. Proc Natl Acad Sci U S A. 2010 Feb 2;107(5):2165–70.
85 Taylor GS, Mautner J, Münz C. Autophagy in herpesvirus immune control and immune escape. Herpesviridae. 2010;2(1):2.
86 Jagannath C, Lindsey DR, Dhandayuthapani S, Xu Y, Hunter RL, Jr., Eissa NT. Autophagy enhances the efficacy of BCG vaccine by increasing peptide presentation in mouse dendritic cells. Nat Med. 2009;15(3):267–76.
87 Russmann H, Panthel K, Kohn B, Jellbauer S, Winter SE, Garbom S, Wolf-Watz H, Hoffmann S, Grauling-Halama S, Geginat G. Alternative endogenous protein processing via an autophagy-dependent pathway compensates for Yersinia-mediated inhibition of endosomal major histocompatibility complex class II antigen presentation. Infect Immun. 2010;78(12):5138–50.
88 Dengjel J, Schoor O, Fischer R, Reich M, Kraus M, Muller M, Kreymborg K, Altenberend F, Brandenburg J, Kalbacher H, Brock R, Driessen C, Rammensee HG, Stevanovic S. Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc Natl Acad Sci U S A. 2005;102:7922–7.
89 Nedjic J, Aichinger M, Emmerich J, Mizushima N, Klein L. Autophagy in thymic epithelium shapes the T-cell repertoire and is essential for tolerance. Nature. 2008;455(7211):396–400.
90 Lee HK, Mattei LM, Steinberg BE, Alberts P, Lee YH, Chervonsky A, Mizushima N, Grinstein S, Iwasaki A. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity. 2010;32(2):227–39.
91 Leib DA, Alexander DE, Cox D, Yin J, Ferguson TA. Interaction of ICP34.5 with Beclin 1 modulates herpes simplex virus type 1 pathogenesis through control of CD4+ T-cell responses. J Virol. 2009;83(23):12164–71.
92 Li Y, Wang LX, Yang G, Hao F, Urba WJ, Hu HM. Efficient cross-presentation depends on autophagy in tumor cells. Cancer Res. 2008;68(17):6889–95.
93 Uhl M, Kepp O, Jusforgues-Saklani H, Vicencio JM, Kroemer G, Albert ML. Autophagy within the antigen donor cell facilitates efficient antigen cross-priming of virus-specific CD8+ T cells. Cell Death Differ. 2009;16(7):991–1005.
94 Pua HH, Dzhagalov I, Chuck M, Mizushima N, He YW. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med. 2007;204(1):25–31.
95 Pua HH, Guo J, Komatsu M, He YW. Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J Immunol. 2009;182(7):4046–55.
96 Jia W, Pua HH, Li QJ, He YW. Autophagy regulates endoplasmic reticulum homeostasis and calcium mobilization in T lymphocytes. J Immunol. 2011;186(3):1564–74.
97 Miller BC, Zhao Z, Stephenson LM, Cadwell K, Pua HH, et al. The autophagy gene ATG5 plays an essential role in B lymphocyte development. Autophagy. 2008;4(3):309–14.
98 Mortensen M, Soilleux EJ, Djordjevic G, Tripp R, Lutteropp M, Sadighi-Akha E, Stranks AJ, Glanville J, Knight S, SE WJ, Kranc KR, Simon AK. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J Exp Med. 2011 Feb 21.
99 Arsov I, Adebayo A, Kucerova-Levisohn M, Haye J, MacNeil M, Papavasiliou FN, Yue Z, Ortiz BD. A role for autophagic protein Beclin 1 early in lymphocyte development. J Immunol. 2011;186(4):2201–9.
100 Arsov I, Li X, Matthews G, Coradin J, Hartmann B, Simon AK, Sealfon SC, Yue Z. BAC-mediated transgenic expression of fluorescent autophagic protein Beclin 1 reveals a role for Beclin 1 in lymphocyte development. Cell Death Differ. 2008;15(9):1385–95.
101 Zhao Z, Fux B, Goodwin M, Dunay IR, Strong D, Miller BC, Cadwell K, Delgado MA, Ponpuak M, Green KG, Schmidt RE, Mizushima N, Deretic V, Sibley LD, Virgin HW. Autophagosome-independent essential function for the autophagy protein Atg5 in cellular immunity to intracellular pathogens. Cell Host Microbe. 2008;4(5):458–69.