Incidence, clinical presentation and imaging findings of cavernous malformations of the CNS
DOI: https://doi.org/10.4414/smw.2011.13172
M
El-Koussy, F
Stepper, A
Spreng, A
Lukes, C
Brekenfeld
Summary
QUESTIONS UNDER STUDY:Up to 88% of cavernous malformations (CMs) of the central nervous system can become symptomatic and cause long-term disability. The aim of this study was to document the characteristics of CMs in the catchment area of our institution.
METHODS:We retrospectively analysed newly discovered CMs over a 20-year observation period, as well as the frequency of familial forms in the catchment area.
RESULTS: In the period from 1985–2004, a total of 347 patients were investigated. The cohort included about 75% symptomatic CM cases. A total of 1.31 and 0.55 symptomatic and asymptomatic cases, respectively, were newly diagnosed per annum per 100’000 inhabitants. Symptomatic CMs were diagnosed on average at the age of 36 years (range: newborn to 79 years old). There were slightly more patients who presented with evidence of acute bleeding (28%) than those with seizures (26%). Most intracranial lesions were supratentorial in location (54%). Lesion size was predominately below 3 cm (range: 0.2 to 8 cm). Symptomatic CMs (average: 1.75 cm) were significantly larger (p <.0001) than asymptomatic ones (average 0.91 cm). When compared to medical literature, there was a relatively high frequency of multiple CMs (18.9%), which were more common in the familial form (62%).
CONCLUSIONS: The observed frequency of CM, including multiple lesions in a single individual and the familial form of this pathological entity appears relatively high compared to medical literature.
Introduction
Cavernous malformations (CM) are so called benign vascular hamartomas, which can be found within the central nervous system (CNS) and cranial nerves [1]. They may grow by intra-lesional haemorrhage, thrombosis and their organisation. Depending on size, location and complications such as major haemorrhage or space occupying effect, the clinical spectrum includes: epileptic seizures, signs of acute intracranial haemorrhage, headaches or symptoms due to space-occupying effect in large lesions. A total of 69–88% of cases become symptomatic at least once. The rest remain clinically asymptomatic for life [2–4]. There are no population based studies. Hsu reported seeing 1.5–2.2 symptomatic patients per year in his neurosurgical clinic [2], while more recent publications report a higher frequency. Porter [4] encountered a total of 173 (symptomatic and asymptomatic) patients between 1989–96 in and around Toronto. The estimated prevalence in the population is about 0.4% [5]. Beside sporadic cases, CM appears as a familial form with autosomal dominant heredity [6].
We aimed to analyse the newly discovered CMs, symptomatic and asymptomatic, over a 20 year observation period, as well as documenting the localisation and size of lesions and reporting the frequency of familial forms in the catchment area of our institution.
Patients and methods
This was an observational, retrospective study of the cavernous malformations in the catchment area of our institution. The Inselspital is a tertiary care centre of Canton Bern and the main teaching hospital of the University of Bern, Switzerland. Our centre covers a catchment area of around 10’000 km2, serving approximately 1.39 M inhabitants. Cases from abroad were excluded. We analysed the medical records of patients who were diagnosed at the Inselspital as having a CNS cavernous malformation between 1985 and 2004. The following clinical data were documented: age, gender, and symptomatic versus asymptomatic lesion. To be defined as “symptomatic” the patient had to present one or a combination of the following clinical symptoms and/or signs; seizure(s), neurological deficit(s), signs of raised intracranial pressure (evidence of mass effect) or an acute attack of headache of previously unknown character. Sporadic recurrent headaches of nonspecific character were not included in this definition. Individuals clinically suspected of having a CM with no previous radiological documentation were contacted and, in case of given consent, underwent a CT or MRI examination.
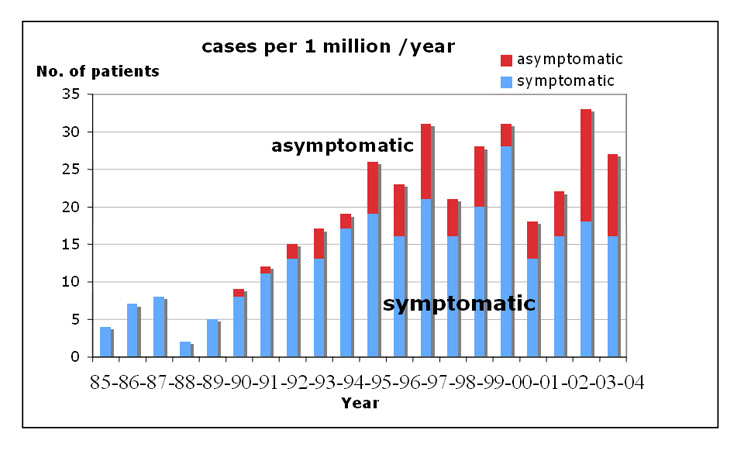
Figure 1
Frequency of newly diagnosed symptomatic and asymptomatic cases from 1985–2004 (cases per 1 million). The figure underscores how problematic the definition of symptomatic versus asymptomatic can be in this topic (the frequency of symptomatic versus asymptomatic cases strongly, possibly artificially, fluctuating over the years).
Radiological points of analysis included: number, location, size of the lesion, intracranial haemorrhage and space occupying effect due to the lesion. A CM presenting due to a recent intracranial haemorrhage was defined with MRI or/and CT signs of acute-to-sub-acute intracerebral (parenchymal) bleeding in or in the vicinity of the lesion, the presence of fluid-fluid level(s) within the CM, or/and a recent subarachnoid or intraventricular haemorrhage, related to the CM. Thus, other causes of haemorrhage, such as arteriovenous malformations or aneurysms, were excluded. The age of the haemorrhage on MRI was interpreted based on the T1- and T2-signal characteristics. On CT, the haemorrhage had to be hyperdense (Hounsfield Units ≥60) to be considered a recent haemorrhage. Chronically bled, calcified lesions and CM with a dark hemosiderin rim on MRI were excluded from the definition of a “recent haemorrhage in the CM”. Pathology reports were included in cases of CMs operated upon.
The search strategy for further affected individuals and family members included examining suspicious history including seizures, focal neurological deficits, stroke-like symptoms and signs of acute intracranial bleeding in the subjects and in first degree relatives younger than 40 years. If suspected, the subjects and their family members were contacted, interviewed in person or by phone, whenever feasible, and, in case of consent, were subjected to a clinical examination to confirm the clinical history, and MRI or CT was performed whenever possible.
Results
Study population
In the period from 1985–2004, 358 patients with CMs of the CNS or optic nerve were investigated at our institution. The patient cohort initially included 147 (41.1%) women and 211 (58.9%) men. Eleven patients were excluded, having been referred from abroad (Libya, Italy, Germany and Russia), mostly in the 1980s. Thus 347 cases were seen from the catchment area of our institution (table 1, fig. 2A).
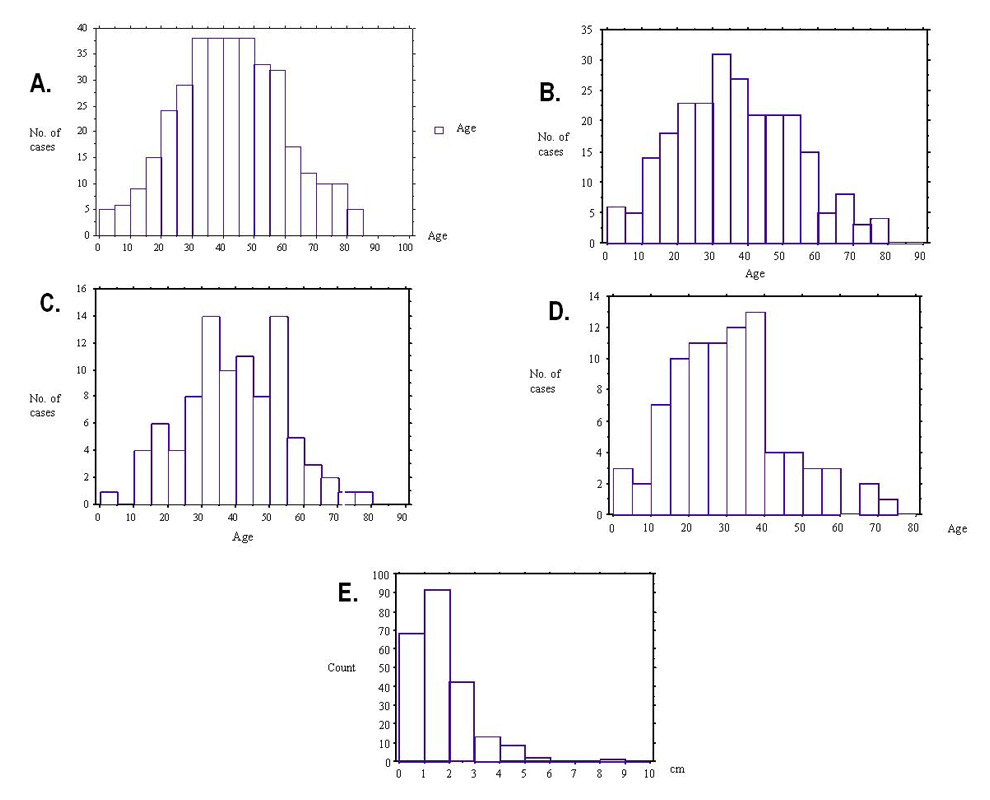
Figure 2
A: Age distribution of cavernous malformations in the studied population
B: Age distribution for the newly diagnosed symptomatic cases
C: Age distribution for haemorrhages due to CMs
D: Age distribution for epilepsy related to CMs
E: Size of CMs in cm (n = 228)
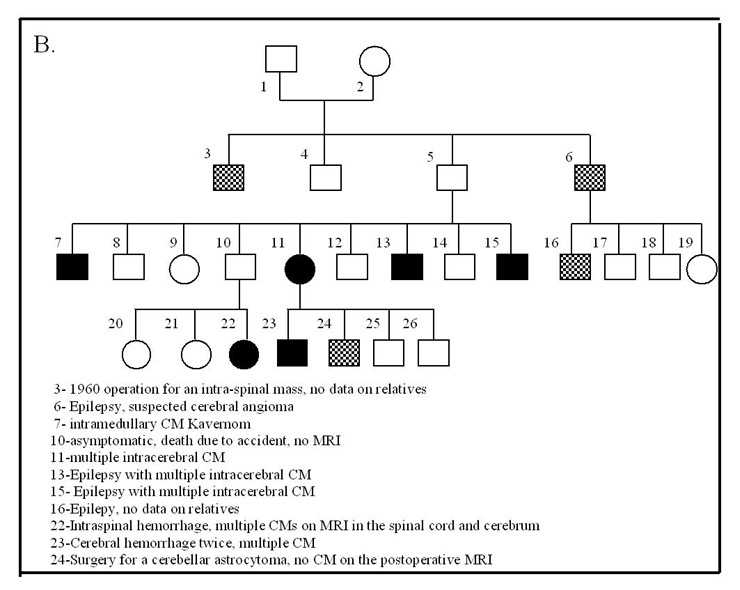
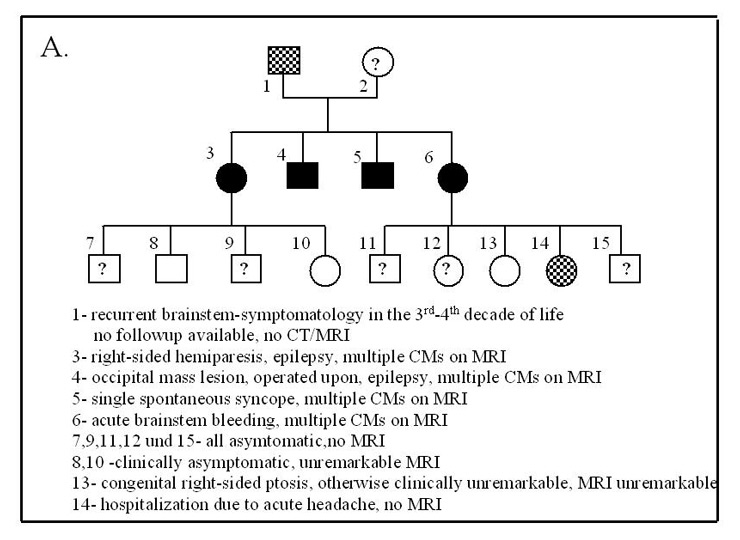
Figure 3
Hereditary CMs.
A: Example Family “S.” with hereditary CMs
B: Example Family “H.” with hereditary CMs
The newly diagnosed cases
The study cohort (n = 347 patients) included 87 (25.07%) asymptomatic and 260 (74.93%) symptomatic CM cases. Till 1995, there was a progressive increase in symptomatic cases accompanied by an increase in asymptomatic patients. The number of the new cases since 1995 remained relatively constant with 13–28 (mean 18.3) registered symptomatic and 5–14 (mean 7.7) asymptomatic cases / year (fig. 1, table 1). Accordingly, an annual incidence of 1.31 and 0.55 newly diagnosed symptomatic and asymptomatic cases respectively per 100’000 inhabitants during the observational period could be calculated.
Clinical findings and age distribution
Acute clinical presentation with focal deficits due to bleeding and seizures were the most common patterns in symptomatic cases (37.3% and 34.6% respectively). Symptoms due to mass effect by the lesions, acute headache and non-specific symptoms accounted for 13.5%, 3.1% and 11.5% respectively (table 2). Symptomatic CMs were diagnosed, on average, at the age of 36 years (range: newborn to age 79 years (fig. 2B)). Asymptomatic patients were, on average, 6.3 years older than symptomatic cases at the time of diagnosis. Radiological evidence of haemorrhages due to a CM was observed at a mean age of 39 years and presented in 95% (88/92 cases) before the age of 66 (fig. 2C). Symptoms / signs of an intra-cranial space-occupying lesion (mean 36 years) and especially seizures (mean 30 years) presented at a slightly younger age (fig. 2D). There was no relevant gender-related difference (p = 0.8).
Number, size and localisation of the lesions
A total of 728 CM lesions were detected with 69% of lesions having a supra-tentorial localisation: 50.3% in a cerebral hemisphere (fig. 4, 6A,B) and 4.2% in the basal ganglia; 22% were infra-tentorial with 14% in the brainstem (fig. 5A) and 8% in the cerebellum; 3% were spinal (fig. 5D) and 1.5% were orbital (fig. 6C,D, table 3). Multiple lesions were detected in 65 patients (18.9%). These cases had an average of 6.9 lesions per patient. The size of lesions could be calculated in 228 patients who were scanned at our institution and was mostly smaller than 3 cm, with a range from 0.2 to 8 cm (fig. 2E). Symptomatic CMs (average diameter: 1.75 cm, n = 164) were significantly larger (p <.0001) than asymptomatic (average diameter 0.91 cm, n = 64).
Aetiology
The exact frequency of the familial type for the whole studied cohort could not be precisely determined since we could not perform systematic family screening. We found 12 families with 29 affected members (8.4% of the studied population) with a typical autosomal dominant pedigree. A total of 18 (62%) of these cases had multiple CMs (fig. 3). An additional 33 patients (9.9% of the cohort) had a family history with intra-cerebral bleeds, seizures and strokes before the age of 40, which pointed to a possible hereditary aetiology. Three patients (0.9%) developed CMs following radiotherapy of brain tumours during childhood and all the CMs appeared within the radiation field. In the remaining patients, the CMs were sporadic / cryptogenic in nature with 90.1% of these cases having a single lesion.
|
Table 1: Number of newly-diagnosed CMs. |
|
Period
|
Total / Symptomatic
|
| 1985–89 |
17 / 17 |
| 1990–94 |
70 / 60 |
| 1995–99 |
129 / 92 |
| 2000–04 |
131 / 91 |
| Total (Percentage) |
347 (100%) / 260 (74.93%) |
|
Table 2:Main symptom / sign and age at the time of presentation (n = 347). |
|
Presenting symptom / sign
|
No. of cases
|
Percentage of total cases (n = 347)
|
Percentage of symptomatic cases
(n = 260)
|
Age at presentation
(mean / StdDev / StdError)
|
| Radiological evidence of recent intracranial haemorrhage |
97 |
28.06% |
37.31% |
38.927 / 15.463 / 1.639 |
| Seizure (s) |
90 |
25.97% |
34.62% |
29.791 / 14.597 / 1.574 |
| Clinical evidence of mass effect |
35 |
10.15% |
13.46% |
36.588 / 20.067 / 3.441 |
| Headache and other symptoms |
38 |
11.05% |
14.62% |
43.194 / 18.615 / 3.102 |
| Asymptomatic |
87 |
24.78% |
n.a. |
n.a. |
| n.a.: not applicable |
|
Table 3:Location and number of the CM lesions according to MRI / CT: (Total number of cases scanned: n = 282). |
|
Lesion location
|
Number of cases
|
Percentage of cases
|
| Cerebral hemispheres |
175 |
50.30% |
| Basal ganglia, thalamus |
15 |
4.19% |
| Brainstem |
49 |
14.07% |
| Cerebellum |
28 |
8.08% |
| Orbital |
5 |
1.50% |
| Spinal canal |
10 |
2.99% |
Discussion
The first case of cavernous vascular growth in the brain was documented in 1854 [7]. The first paper on the potentially hereditary nature of the illness (heredofamilial angiomatosis) was published in 1928 [8]. Cavernous malformations are single or multiple mulberry-like masses of closely apposed immature blood vessels (“caverns”) lined by a thin endothelium, and lacking muscle cells, elastic fibres and intervening parenchyma. Usually no sizeable feeding or draining vessels are located within or in close vicinity to a CM, making it a low-flow “angioma” often occult on conventional angiography [9]. Acute or organised thrombi are usually found in CMs. Chronic small bleeds lead to a gliotic layer stained with haemosiderin around the lesion. Cavernous malformations are characteristically “dynamic” [10]. Recurrent haemorrhages and thromboses as well as cyst formation in a lesion can lead to lesion growth, with a size up to 10 cm, and may cause pressure on the surrounding brain parenchyma [11–13]. They may also shrink significantly but rarely remain quiescent. Rare subtypes include the cystic type, the extra-axial type (fig. 5B) and the calcified haemangioma type [14].
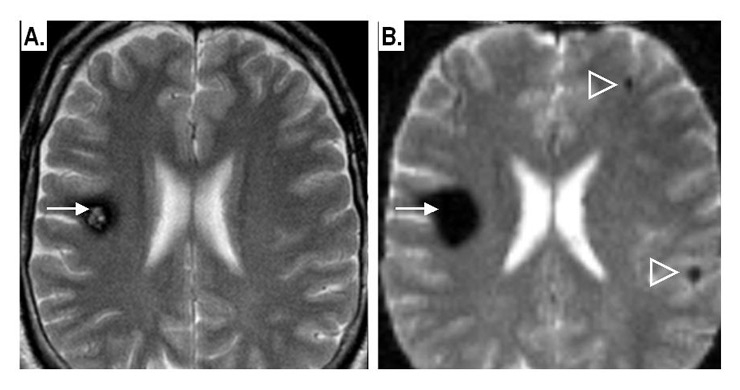
Figure 4
Example of supra-tentorial intra-parenchymal CMs:
A: conventional T2-weighted spin-echo MRI image: shows a solitary CM (arrow) in the right frontal lobe. The lesion shows a “pop-corn” appearance surrounded by a dark rim of haemosiderin.
B: Same patient, same axial section, T2-weighted gradient-echo, echoplanar image: notice the “blooming effect” due to susceptibility artefacts by the CM-related haemosiderin, with two small lesions now detectable in the contralateral hemisphere (arrowheads).
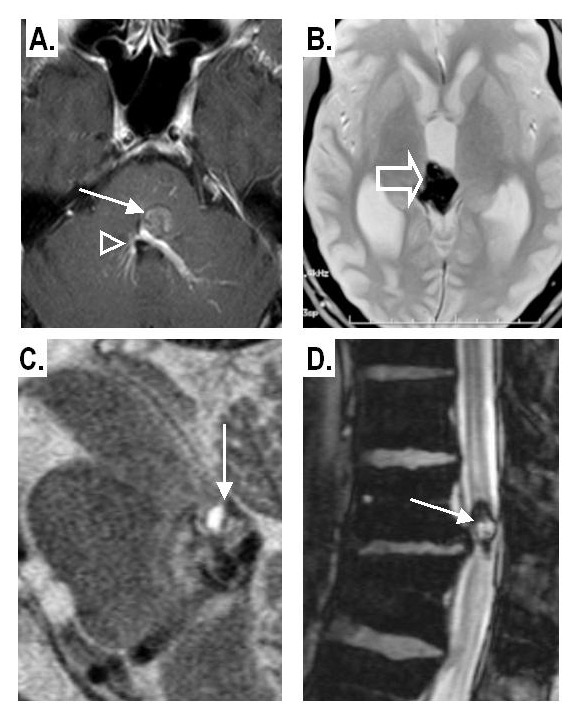
Figure 5
A: Co-existence of a CM in the brainstem (arrow) with a developmental venous anomaly (arrowheads) in the contrast-enhanced T1-weighted image.
B: Intra-ventricular CM: within the third ventricle well seen on a gradient-echo T2* image.
C: Infra-tentorial CM with a “fluid-fluid level” pattern on a sagittal T2-weighted spin-echo image
D: Intra-spinal CM; dark haemosiderin rim on gradient-echo T2* sequence
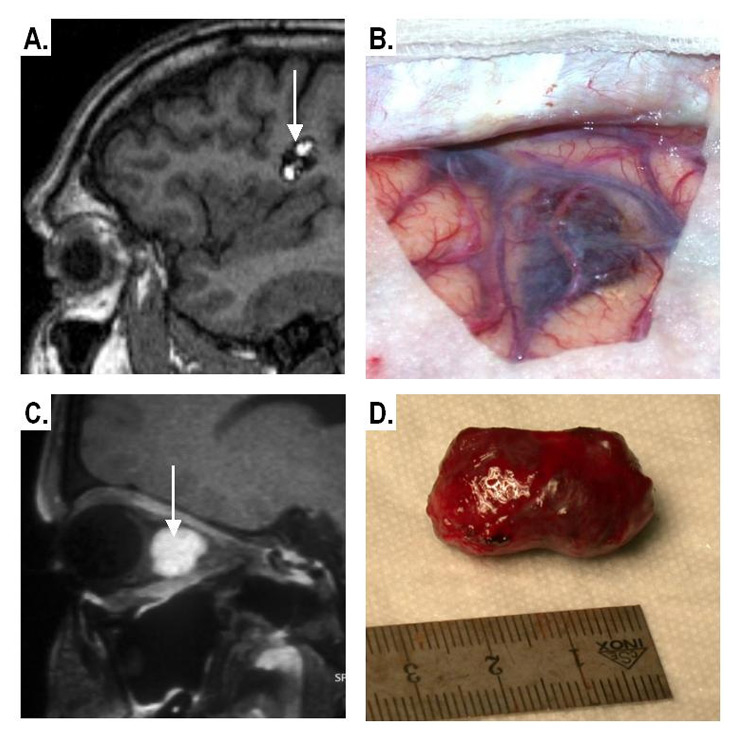
Figure 6
Imaging and gross pathological appearance of CM.
Supra-tentorial CM
A: Non-contrast T1-weighted MRI showing the typical “popcorn-like” appearance
B: Intra-operative image
Intra-orbital CM
C:Contrast-enhanced fat-suppressed MRI (well-defined enhancing intraconal mass).
D: Gross pathology of the CM after excision.
The CMs of the CNS occur predominantly as single lesions. In 9–18.7% of the affected persons, multiple lesions can be detected [4, 5, 15]. In our series 18.9% of subjects had multiple lesions, thus lying in the upper end of the previously published range.
CMs of the CNS were considered a rare entity, masquerading under their clinical sequelae. In 1928, 44 cases were reported in the world literature [16], in 1976 there were 164 cases [17] and in 1999, 1472 cases were published [18]. A prevalence of 0.02% at autopsy was initially ascertained [19].With the introduction of computed tomography (CT) in the late 1970s and magnetic resonance imaging (MRI) in the late 1980s, CMs were found increasingly more frequently. In Bern, there was a continuous rise of newly diagnosed cases until 1995 (coinciding with the evolution of the “MRI era”). The asymptomatic cases were discovered later than the symptomatic ones. In the SIVMS study from Scotland, the crude detection rate (per 100’000 adults per year) for CMs was 0.56 versus 2.27, 1.12, 0.43 and 0.16 for all intracranial vascular malformations, arteriovenous malformations (AVMs), venous malformations and dural AVMs respectively [20]. In our series, the rate was 1.31 for symptomatic and 0.55 for asymptomatic cases (per 100’000 cases per year). Although not totally comparable (our study included all age-groups, while SIVMS included subjects aged ≥16 y), this series showed a higher incidence rate than in SIVMS. One of the explanations may be the high density of MRI scanners in our region and the special interest in the topic. Previous large series revealed no gender dominance [2], and this result was confirmed by the present study. However, it was previously found that men show earlier and more frequent seizures, while women became symptomatic later and manifest mainly by focal neurological deficits or signs due to larger bleedings [21, 22]. We found no significant gender-related difference in symptomatology.
In our study, around 75% of cases were symptomatic which is in accordance with previous figures of 69–88% of cases with CMs becoming symptomatic at least once in their life [2–4]. The lesions had a supra-tentorial location in 74% in previous studies [2] and in 54.5% in our sample. Isolated infra-tentorial CMs were less common (22.15%), while spinal and orbital lesions were quite rare in our study (3% and 1.5% respectively). The MRI appearance of the orbital CMs in our series (fig. 6C,D) was similar to that described previously by Hejazi and co-workers, who suggested that orbital CMs represent a distinct clinicopathologic and neuroradiologic entity. They found that orbital CMs, in comparison with cerebral CMs, showed non-degenerated well-developed vessel walls and were covered by a hard and compact capsule. On MRI these lesions were well defined, exhibited a homogeneous MRI-signal and enhancement patterns. Calcification was rather rare [23].
CT has a sensitivity of 36–74% [21, 24] to detect CMs by exhibiting a space-occupying effect, focal nodular hyperdensity and calcifications on unenhanced CT and mild enhancement on CT after intravenous iodinated agents [25]. MRI remains the method of choice for the detection of CMs. The signal behaviour is determined by the stage of bleeding or thrombus and thus the haemoglobin form in the lesion. Characteristic appearances include the “popcorn” as well as the rare “fluid-fluid level” pattern (fig. 5C). The susceptibility-weighted MRI uses the “blooming effect” due to the susceptibility effects of the blood by-products to enhance lesion detectability (fig. 4B). CMs present with variable degrees of contrast enhancement and, thus, are definitely not distinguishable from AVM when assessed with contrast-enhanced MRI (fig. 5A) [26]. A classification of CMs based on MRI was originally proposed by Zabramski [27, 28].
The clinical features of CMs depend on the lesions’ number, size, location, complication such as haemorrhage, and the age of the patient. It has to be noted that evidence of acute intracranial bleeding, seizures, space-occupying effect and acute headache are not mutually exclusive clinical presentations in cavernous malformations and may coexist in a single case. Seizures are the dominant symptom in 36–39% of patients with most lesions seen in the frontal and temporal lobe [29]. CMs have a high epileptogenic potential attributed to irritation of the adjacent parenchyma by the “haemosiderin fringe” [11].A space-occupying effect, signs of acute haemorrhage, and headache are dominant in 20–32%, 19–25% and 6–8% of cases respectively. In the cohort from Bern, the symptomatology was most common due to acute bleeding (37.3% of symptomatic cases) and seizures (34.6%), followed by mass effect (13.5%) and headache (3.1%). Asymptomatic cases were more frequent (24.8%) than reported in the literature (12–21%) [2, 4].
Haemorrhage due to a CM is usually intra-parenchymal and rarely subarachnoid or intraventricular. MRI criteria for acute intra-cerebral CM-related bleeding include: signal changes corresponding to acute/sub-acute bleeding outside the haemosiderin-ring of the CM, blood on lumbar puncture and autopsy or surgically proven cases [30].Catastrophic consequences of acute bleeding are exceptional due to the low intra-lesional pressure. Mortality is usually low, and is highest (20%) for brainstem CMs with haemorrhage [31]. Morbidity, however, is significant, since only 1/3 of acutely symptomatic patients recover completely, while another third shows no considerable recovery [32].
Usually CMs become symptomatic with a size above 1 cm in diameter [2] and during the 2nd–4th decade of life with an average age of 37.5 years [4, 22]. In this series, most cases presented during the fourth decade of life with the age distribution having a near normal Gaussian pattern (fig. 2). Patients presenting with seizures were, on average, younger (average: 29.8 y) than those with evidence of acute intra-cranial haemorrhage (average: 38.9 y) or mass effect (average: 36.6 y). Lesion size in our sample varied (0.2–8 cm; 90% <3 cm) and was double in symptomatic (average: 1.75 cm) compared to asymptomatic (average: 0.91 cm) patients.
Familial forms with autosomal dominant transmission were initially described in the 1920s [8] and have been confirmed by genetical analyses in 1994 [33]. In the pre-MRI era, 6 families were known [34–36]. With the introduction of MRI, more families were discovered [37] and thus in 1998 109 families were known, and in the USA most families were of Mexican descent [24]. The frequency of the hereditary form in the CM-affected population is 6–50% [2, 24, 37]. In the cohort from Bern, 8.4% of the studied population had the familial “autosomal dominant form”, which was documented in 29 cases from 12 families. The incidence of multiple CMs in the subjects with the hereditary form (62%) was higher than in the whole cohort (18.9%) and thus higher than that for the sporadic form. This confirms the report from Zamaki et al. who observed that 84% of the affected family members had multiple (average 6.5) lesions [28]. This is in contrast to sporadic cases, which show mostly a single CM. The radiological penetrance for the familial variant is usually complete by the age of 45. The symptom penetrance with the hereditary form is 54–71% [3, 6, 28]. The prevalence of symptomatic cases in a study with hereditary cases amounted to 54% of the affected members of the family at the time of the investigation [6, 38].
According to current literature, a familial form of CM should be suspected in cases with multiple CMs and/or a family history with epilepsy, strokes, focal neurological deficits and signs of intracranial bleeding in first degree relatives younger than 40 years [39]. In the current study, an additional 9.9% of the cohort could have some type of hereditary form based on the aforementioned family history criteria. With such patients it is recommended to examine all first-degree relatives with MRI. A hereditary form is defined if more than one person in a family is affected. A person within an affected family is considered not affected if the head-MRI after the 20th year of life is unremarkable. CMs have been also linked to cranial radiation in childhood [40], which was the case in three of our patients.
Our study suffers several limitations; we retrospectively analysed for individuals with CMs of the CNS who presented to our institution in the study period and we screened for their families. Thus, the screening in the catchment area was not totally systematic (as we mentioned previously and as supported by table 1) and it may well be that a number of CM patients from the catchment area were not diagnosed at our institution and thus not included in this study. This might be one of the explanations for the rather high incidence of CMs observed in our study.
Summary
With an incidence of 1.31 newly diagnosed cases / 100’000 inhabitants / year in the catchment area of the University Hospital Bern, symptomatic CMs were more frequent than in some European countries. Still this frequency does not justify systematic screening for CMs. Our patients presented with first symptoms at a more advanced age (average 36 years) than previously reported. Slightly more patients presented with evidence of acute bleeding (28%) than with seizures (26%). The opposite was found in previously published studies. Patients from our centre had a high frequency of multiple CMs (18.9%). Although the incidence of multiple CMs was higher in patients with the familial form (62%) compared to the sporadic form, this was still lower than that reported previously.
Correspondence:
Marwan El-Koussy MD
Institute of Diagnostic and Interventional Neuroradiology
Inselspital
University of Bern
Freiburgstrasse 4
CH-3010 Bern
Switzerland
marwan.el-koussy@insel.ch
References
1 Zentner J, Hassler W, Gawehn J, Schroth G. Intramedullary cavernous angiomas. Surg Neurol. 1989;31:64–8.
2 Hsu F, Rigamonti D, Huhn SL. Epidemiology of Cavernous Malformations. In: Awad IA, Barrow DL, eds. Cavernous Malformations: AANS Publications Committee; 1993:13–23.
3 Polymeropoulos MH, Hurko O, Hsu F, et al. Linkage of the locus for cerebral cavernous hemangiomas to human chromosome 7q in four families of Mexican-American descent. Neurology. 1997;48:752–7.
4 Porter PJ, Willinsky RA, Harper W, Wallace MC. Cerebral cavernous malformations: natural history and prognosis after clinical deterioration with or without hemorrhage. J Neurosurg. 1997;87:190–7.
5 Otten P, Pizzolato GP, Rilliet B, Berney J. 131 cases of cavernous angioma (cavernomas) of the CNS, discovered by retrospective analysis of 24535 autopsies. Neurochirurgie. 1989;35:82–3,128–31.
6 Gunel M, Awad IA, Finberg K, et al. Genetic heterogeneity of inherited cerebral cavernous malformation. Neurosurgery. 1996;38:1265–71.
7 Luschka H. Kavernöse Blutgeschwulst des Gehirns. Virchows Arch path Anat. 1854;6:458–70.
8 Kufs H. Über heredofamiliäre Angiomatose des Gehirns und der Retina, ihre Beziehung zueinander und zur Angiomatose der Haut. Z Neuol Psychol. 1928;113:651–86.
9 Little JR, Awad IA, Jones SC, Ebrahim ZY. Vascular pressures and cortical blood flow in cavernous angioma of the brain. J Neurosurg. 1990;73:555–9.
10 Labauge P, Brunereau L, Levy C, Laberge S, Houtteville JP. The natural history of familial cerebral cavernomas: a retrospective MRI study of 40 patients. Neuroradiology. 2000;42:327–32.
11 Johnson PC, Wascher TM, Golfinos J, Spetzler RF. Definition and pathological features. In: Awad IA, Barrow DL, eds. Cavernous Malformations: American Association of Neurological Surgeons, Publications Committee; 1993:1–11.
12 Steiger HJ, Markwalder RV, Reulen HJ. Cerebral cavernoma as a cause of recurrent cerebral hemorrhage and epileptic insults. Schweiz Med Wochenschr. 1988;118:471–7.
13 van Lindert EJ, Tan TC, Grotenhuis JA, Wesseling P. Giant cavernous hemangiomas: report of three cases. Neurosurg Rev. 2007;30:83–92; discussion.
14 Zabramski JM, Awasthi D. Extra-axial cavernous malformations. In: Awad IA, Barrow DL, eds. Cavernous Malformations: American Association of Neurological Surgeons, Publications Committee; 1993:133–44.
15 Del Curling O, Jr., Kelly DL, Jr., Elster AD, Craven TE. An analysis of the natural history of cavernous angiomas. J Neurosurg. 1991;75:702–8.
16 Dandy WE. Venous abnormalities and angiomas of the brain. Arch Surg. 1928;11:715–93.
17 Voigt K, Yasargil MG. Cerebral cavernous haemangiomas or cavernomas. Incidence, pathology, localization, diagnosis, clinical features and treatment. Review of the literature and report of an unusual case. Neurochirurgia. (Stuttg) 1976;19:59–68.
18 Hahn M. CM des zentralen Nervensystems. 111 eigene Fälle und Metaanalyse von 1361 Literaturfällen. Medical thesis. Heidelberg, Germany: University of Heidelberg, Germany; 1999.
19 Berry RG, Alpers BJ, White JC. The site, structure and frequency of intracranial aneurysms, angiomas and arteriovenous abnormalities. Res Publ Assoc Res Nerv Ment Dis. 1966;41:40–72.
20 Al-Shahi R, Bhattacharya JJ, Currie DG, et al. Prospective, population-based detection of intracranial vascular malformations in adults: the Scottish Intracranial Vascular Malformation Study (SIVMS). Stroke. 2003;34:1163–9.
21 Requena I, Arias M, Lopez-Ibor L, et al. Cavernomas of the central nervous system: clinical and neuroimaging manifestations in 47 patients. J Neurol Neurosurg Psychiatry. 1991;54:590–4.
22 Robinson JR, Awad IA. Clinical spectrum and natural course. In: Awad IA, Barrow DL, eds. Cavernous Malformations: American Association of Neurological Surgeons, Publications Committee; 1993:25–36.
23 Hejazi N, Hassler W, Offner F, Schuster A. Cavernous malformations of the orbit: a distinct entity? A review of own experiences. Neurosurg Rev. 2007;30:50–4; discussion 4–5.
24 Rigamonti D, Hadley MN, Drayer BP, et al. Cerebral cavernous malformations. Incidence and familial occurrence. N Engl J Med. 1988;319:343–7.
25 Lobato RD, Perez C, Rivas JJ, Cordobes F. Clinical, radiological, and pathological spectrum of angiographically occult intracranial vascular malformations. Analysis of 21 cases and review of the literature. J Neurosurg. 1988;68:518–31.
26 Pinker K, Stavrou I, Knosp E, Trattnig S. Are cerebral cavernomas truly nonenhancing lesions and thereby distinguishable from arteriovenous malformations? MRI findings and histopathological correlation. Magn Reson Imaging. 2006;24:631–7.
27 Raychaudhuri R, Batjer HH, Awad IA. Intracranial cavernous angioma: a practical review of clinical and biological aspects. Surg Neurol. 2005;63:319-28; discussion 28.
28 Zabramski JM, Wascher TM, Spetzler RF, et al. The natural history of familial cavernous malformations: results of an ongoing study. J Neurosurg. 1994;80:422–32.
29 Simard JM, Garcia-Bengochea F, Ballinger WE, Jr., Mickle JP, Quisling RG. Cavernous angioma: a review of 126 collected and 12 new clinical cases. Neurosurgery. 1986;18:162–72.
30 Robinson JR, Awad IA, Little JR. Natural history of the cavernous angioma. J Neurosurg. 1991;75:709–14.
31 Fritschi JA, Reulen HJ, Spetzler RF, Zabramski JM. Cavernous malformations of the brain stem. A review of 139 cases. Acta Neurochir. (Wien) 1994;130:35–46.
32 Sage MR, Brophy BP, Sweeney C, et al. Cavernous haemangiomas (angiomas) of the brain: clinically significant lesions. Australas Radiol. 1993;37:147–55.
33 McKusick VA. Mendelian inheritance in man: a catalog of human genes and genetic disorders. Baltimore: John Hopkins University Press; 1994.
34 Bicknell JM, Carlow TJ, Kornfeld M, Stovring J, Turner P. Familial cavernous angiomas. Arch Neurol. 1978;35:746–9.
35 Clark JV. Familial occurrence of cavernous angiomata of the brain. J Neurol Neurosurg Psychiatry. 1970;33:871–6.
36 Hayman LA, Evans RA, Ferrell RE, Fahr LM, Ostrow P, Riccardi VM. Familial cavernous angiomas: natural history and genetic study over a 5-year period. Am J Med Genet. 1982;11:147–60.
37 Steichen-Gersdorf E, Felber S, Fuchs W, Russeger L, Twerdy K. Familial cavernous angiomas of the brain: observations in a four generation family. Eur J Pediatr. 1992;151:861–3.
38 Gunel M, Awad IA, Finberg K, et al. A founder mutation as a cause of cerebral cavernous malformation in Hispanic Americans. N Engl J Med. 1996;334:946–51.
39 Dellemijn PL, Vanneste JA. Cavernous angiomatosis of the central nervous system: usefulness of screening the family. Acta Neurol Scand. 1993;88:259–63.
40 Novelli PM, Reigel DH, Langham Gleason P, Yunis E. Multiple cavernous angiomas after high-dose whole-brain radiation therapy. Pediatr Neurosurg. 1997;26:322–5.