Mechanisms of aneuploidy and its suppression by tumour suppressor proteins
DOI: https://doi.org/10.4414/smw.2011.13170
C
Thoma, A
Toso, P
Meraldi, W
Krek
Summary
Aneuploidy, as a result of numerical changes in chromosome number, was observed in tumours almost a century ago. The molecular mechanisms underlying this phenomenon and their impact on tumour development are still poorly understood. A series of recent observations provide direct linkages between the normal function of tumour suppressor proteins and the suppression of aneuploidy. The prospects that these findings offer for understanding the role of aneuploidy in cancer are discussed in this review.
Introduction
The term cancer is used to describe a collection of genetic diseases that allow inappropriate cell proliferation within the body. Despite the wide variety of genetic alterations that lead to different pathological manifestations among these diseases, most cancers share common underlying hallmarks contributing to a tumourigenic phenotype such as evasion of apoptosis, self-sufficiency in growth signals, insensitivity to anti-growth signals, sustained angiogenesis, limitless replicative potential, tissue invasion & metastasis, evasion of immune surveillance [1, 2] and the stress phenotypes of cancer, including metabolic, proteotoxic, mitotic, oxidative and DNA damage stress [3]. Cancer cells often gain these phenotypic alterations by gain-of-function mutation, amplification and over-expression of oncogenes together with loss-of-function mutation, deletion and epigenetic silencing of tumour suppressor genes [4]. Although tumourigenesis in humans is a multi-step mutagenic process, a number of key mutations in certain cancer types are predominant. These mutations result in the activation of particular oncogenes with dominant gain of function and inactivation of tumour-suppressor genes with recessive loss-of-function phenotypes characteristic for a certain cancer. For example, high frequencies of mutations of the tumour suppressor genes adenomatous polyposis coli (APC) or von Hippel-Lindau (VHL) are detected in the large and small intestine and kidney and adrenal glands, respectively [5]. Together with an accumulation of other somatic mutations, this ultimately promotes tumour cell evolution. Intriguingly, development of cancer is often accompanied by an alteration in number of whole chromosomes called aneuploidy, caused by a process known as chromosome instability (CIN). The notion that aneuploidy could be a causal event in the process of tumourigenesis was already proposed by Theodor Boveri [6]. Therefore, understanding the detailed mechanisms of how aneuploidy contributes to development, progression or even suppression of cancer and how tumour suppressor gene mutations contribute to CIN remains a major challenge.
Aneuploidy, a result of chromosomal instability in cancer cells
Aneuploidy is a phenomenon observed in over 90% of all solid tumours. Even haematological cancers, known to have a rather stable chromosome number, frequently display loss or gain of few chromosomes [7]. How aneuploidy contributes to cancer development is highly debated and the opinions diverge from being a primary cause of tumourigenesis [8] to being solely a benign epiphenomenon accompanying neoplastic growth [9]. In addition, aneuploidy could even have, under certain circumstances, a tumour suppressive effect [10]. Tumour cells mostly become aneuploid due to chromosome instability (CIN). In this context it is important to distinguish between numerical CIN and structural CIN. Numerical CIN referred as whole CIN (W-CIN) is characterised by the gain or loss of whole chromosomes during cell division, whereas structural CIN (S-CIN) is characterised by structural changes of chromosomes by gain, loss or translocation of chromosome fragments mainly caused by breakage and inappropriate rejoining of these fragments during the process of DNA replication [11]. W-CIN and S-CIN together contribute to three phenomena known to drive cancer: Loss of heterozygosity (LOH) of tumour suppressor genes, enhanced copy number of oncogenes and creation of oncogenic fusion products. The latter can only arise due to S-CIN, as for example the BCR-ABL gene fusion product of parts of chromosome 9 and 22 resulting in the “Philadelphia chromosome”, the main driver in chronic myeloid leukaemia [12]. Chromosomal translocations also occur in solid tumours often at an early stage, these specific and characteristic changes already in the benign state often serve as a diagnostic marker [13]. These abnormalities are often the cause of an impaired double-strand break repair system or telomere maintenance system. In this review, we focus specifically on the molecular mechanisms of W-CIN, how W-CIN arises and we discuss specifically the role of tumour suppressor mutations as a driving mechanism of W-CIN.
Mechanisms leading to whole-chromosome instability
Defects in two distinct processes are considered to be the main causes for W-CIN and include (i) a failure in the centrosome-duplication cycle leading to multiple centrosomes (fig. 1A) and (ii) a dysregulation of the cell division control machinery resulting in lagging chromosomes mainly elicited by a weakened or an over-activated mitotic checkpoint also known as spindle assembly checkpoint (SAC) (fig. 1B).
The generation of multiple centrosomes is known to lead to multipolar spindles followed by cell death [14]. However, when centrosomes are clustered into two poles, merotelic MT-kinetochore attachments can occur: that single kinetochore is attached to MTs emanating from both poles and although erroneous this structure still fulfils the SAC. Such errors in the process of the centrosome duplication cycle occur also under normal conditions but they are corrected by a dedicated control machinery involving Aurora kinase B [15, 16]. Irrespective, when clustering of multiple centrosomes occurs it can cause an overloading of the correction mechanism eventually resulting in W-CIN (fig. 1B) [17]. In contrast to the mechanisms suppressing the generation of multiple centrosomes, the mitotic checkpoint ensures during cell division the correct distribution of the duplicated chromatids into each daughter cell [18]. Two important components are needed to ensure error-free completion of this task: the mechanical machinery and a switch-like control process, the mitotic checkpoint. The mitotic spindle consists of two separate poles built by the centrosomes, where microtubule polymerisation is induced to build the mitotic spindle. A specific subset of these microtubules the kinetochore microtubules (k-MT), are needed to attach the chromosomes via their kinetochores to the adjacent spindle poles. It is exactly this huge multiprotein complex, that builds the interface between the chromosomes and the mechanical part, the mitotic spindle, and it is therefore ideally suited to function as a hub for the SAC [19]. As long as chromosomes are not correctly attached from both spindle poles via k-MTs a STOP signal is produced at the kinetochore. Under conditions where the mitotic checkpoint is fully operational one single unattached kinetochore is sufficient to delay progression to anaphase [20]. This delay is mediated by inhibition of the anaphase promoting complex/cyclosome (APC/C) mainly by one complex localised at the kinetochore: the mitotic arrest deficient homolog (Mad)1-Mad2 catalytic checkpoint complex [21]. Whereas the importance of the adjoining kinetochore complex composed of the proteins budding inhibited by benzimidazole (Bub)R1-Bub3-CENP (centromeric protein)-E lies mainly in controlling the correct tethering between k-MTs and kinetochores (fig. 1B) [22, 23]. Furthermore, the kinetochore localisation of some of the above mentioned components from these two complexes depend on Bub1 [24], although this recruiting function is not entirely clear, Bub1 has an important function in SAC fidelity [25]. The correct and timely regulated assembly of these proteins at the unattached kinetochore leads finally to the generation of a diffusible Mad2 STOP-signal that prevents onset of anaphase.
The Mad1-Mad2 catalytic complex [26] at unattached kinetochores converts free-diffusible Mad2 from an open conformation (O-Mad2) into a closed conformation (C-Mad2) [27]which then in turn sequesters Cdc20 [28] and prevents it from activating the APC/C. Upon correct bipolar tethering of the k-MTs to the two kinetochores of one chromosome with the help of the “fishing-rod like” mitotic kinesin CENP-E [29, 30]. The active Mad1-Mad2 catalytical complex is stripped from the attached kinetochore in a dynein dependent manner and its converting activity is therefore lost [31, 32]. The available amount of free-diffusible Mad2 seems to be crucial and any imbalance in the levels leads to a change in the STOP-signal strength, from either too weak or too strong. Defects in both directions lead to aneuploidy, as it has been shown in cell culture as well as in mouse models [33, 34]. These observations show that Mad2 protein levels have to be tightly controlled and as such represent an “Achilles heel” of the SAC in aneuploidy suppression.
The mitotic checkpoint and cancer
Despite the identification of key mitotic checkpoint proteins, mutations in its components are rarely found in cancer [35, 36]. Moreover, studies in mice have shown that complete knock-outs of many mitotic checkpoint components (e.g., Bub1, Bub3, BubR1, CENP-E, Mad1 and Mad2) are embryonically lethal [37–42]. Indeed a complete loss of the mitotic checkpoint induces cell-intrinsic mitotic cell death or cell death in the ensuing interphase. In accordance with this observation and the importance of the mitotic checkpoint, dysregulation instead of full deactivation of mitotic checkpoint components is more frequently observed in many cancers [43]. Many mouse models have been developed, where mitotic checkpoint components were deleted, reduced or over-expressed and provided significant insight into the complex relationship between aneuploidy and tumour development. Specifically mouse models with either reduced or over-expressed Bub1 [39], Bub3 [40, 44], BubR1 [45, 46], CENP-E [10, 42], Mad1 [38] and Mad2 [33, 34, 37] have been generated. Although all of these checkpoint proteins appear to be essential for viability and checkpoint function, mice do not develop spontaneous tumours in all cases. For example Bub3 heterozygous mice do not develop tumours [44], but a higher incidence of tumours in the lung are observed following treatment of mice with DMBA (9,10-Dimethyl-1,2-benzanthracene, a chemical mutagen) [47]. Also mice heterozygous or hypomorphic for BubR1 fail to develop spontaneously tumours although they also show an increased tumour incidence upon DMBA induction [46]. This implies that additional stress signals are required to elicit tumour development in the face of alterations in mitotic checkpoint protein expression.
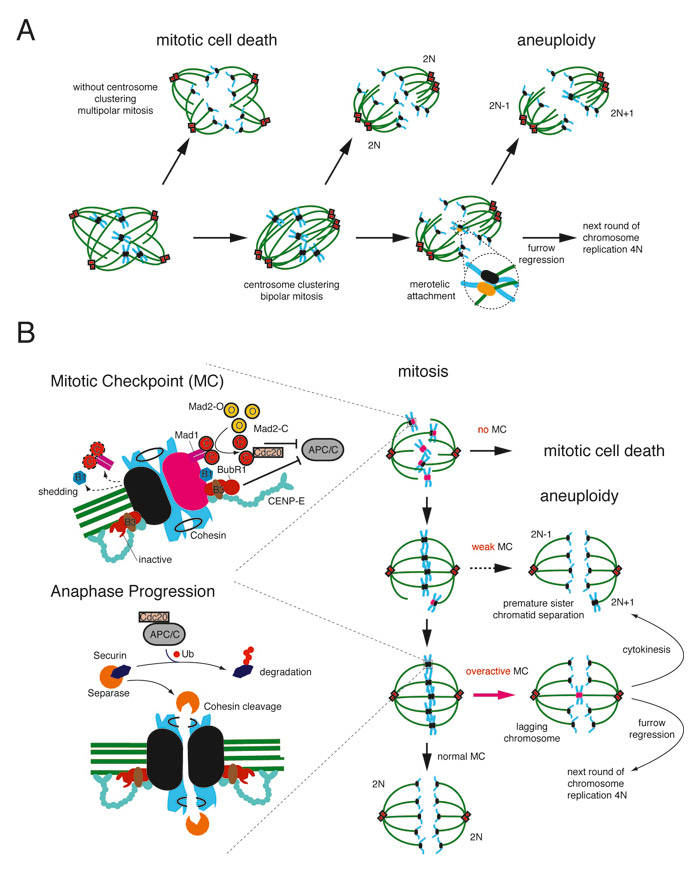
Figure 1
A) Schematic drawing how multiple centrosomes may lead to whole chromosome instability or mitotic cell death. Centrosomes (red), microtubules (green), chromosomes (blue), kinetochores (black), merotelic attached kinetochore (yellow), 2N = diploid chromosome content.
B) Schematic drawing of the mitotic checkpoint and the consequences of failure during mitosis. Left, top: zoom of the mitotic checkpoint components on the kinetochore either inactivated with attached microtubules (black) or activated without any attached microtubules (pink). Left, bottom: zoomed view of one kinetochore with correct bipolar attachment of microtubules exemplifying the fulfilment of the mitotic checkpoint when all kinetochores are attached correctly to micortubules and the molecular consequences for progression to anaphase. Right, top – bottom: consequences in mitosis due to an aberrant mitotic checkpoint leading either to mitotic cell death, aneuploidy or furrow regression. Mad1: mitotic-arrest deficient 1, Mad2-C: mitotic-arrest deficient 2 in a closed conformation, Mad2-O: mitotic-arrest deficient 2 in an open conformation, BubR1: budding inhibited by benzimidazole (Bub) R1, CENP-E: centromeric protein E, APC/C: anaphase promoting complex/ cyclosome, Cdc20: cell diviosion cycle 20, Ub: ubiquitin.
In contrast, mouse models hypomorphic for Bub1[39], heterozygous for CENP-E[10], heterozygous for Mad1 [38], heterozygous for Mad2 [33, 37] or overexpressing Mad2 [34], all spontaneously develop tumours preferentially in the lung, with an incidence of 20–50% but at a high latency of about 20 months [48, 49]. Two important questions arise from these studies. First, what is the reason for this high latency time for tumour formation and why is tumour development restricted to certain organs, particularly the lung and second, why are certain SAC components cancer prone and others not.
Aneuploidy as an elicitor of a stress response and accelerator of cellular evolution
The first studies linking aneuploidy to the fitness of mammalian cells were conducted in primary fibroblasts of trisomy 21 patients. It was shown that these cells harbouring an extra chromosome 21 proliferate more slowly than euploid control cells [50]. Studies in yeast strains engineered to carry one or two extra chromosomes revealed similar defects in cell proliferation. Interestingly, these strains exhibited also a characteristic stress response gene expression pattern [51]. In an elegant experiment, trisomic MEFs were established harboring an extra chromosome 1, 13, 16, or 19. Analysis of these aneuploid cells revealed again a reduced proliferation rate, independent on the chromosome causing the trisomy. In addition metabolic changes were also observed [52]. Based on these observation, it has been proposed that probably an imbalance in the cellular protein composition and consequently an elicited stress response underlies these shared phenotypes of having an extra chromosome [53]. Therefore mutations eliciting W-CIN destabilise the karyotype of a cell and the resulting mitotic and proteotoxic stress could strongly drive a selection for mutations that protect cells from aneuploidy-reduced fitness reduction and towards tumourigenesis [54] at least in yeast cells [55]. Aneuploidy as a driver of tumour evolution by exhibiting a “mutator” phenotype, which is a driving force and accelerator for the acquisition of further cancer-promoting mutations, would further provide an explanation for the late occurrence of tumours in the above discussed mouse models of the SAC as well as why mutagenic agents induce or accelerate tumour formation in these mice [48]. Indeed it seems that aneuploidy itself is not sufficient to induce tumour formation, rather that additional cellular functions may have to be impaired for cellular transformation. A possible reason why an imbalance of certain mammalian SAC components is cancer prone and an imbalance of others is not is that the cancer-prone SAC components could have additional cellular functions besides SAC regulation.
In a recent report it was shown that CIN elicited by overexpression of Mad2 in a K-Ras-driven lung cancer model is not sufficient to overcome oncogene-addiction but leads to accelerated tumour relapse after oncogene withdrawal [56]. This indicates that the combination of a chromosomally unstable setting together with a deregulation of key cellular pathways is initially important but recurrence can occur with emergent independence from the original oncogenic stimulus probably driven by the evolutionary mutagenic fuel provided by CIN. Interestingly the loss of some well-characterised tumour suppressor proteins couples indeed a deregulation of key cellular pathways with karyotypic imbalance. This creates a double burden on a cell harbouring such tumour suppressor protein inactivating mutations with significant implications for tumour development.
The role of tumour suppressor proteins in the suppression of aneuploidy
Several lines of evidence show that the deregulation of key signalling pathways in combination with CIN drives at least some aspects of tumour initiation, development or relapse after therapy. Deregulation of key signalling pathways most often is a cause of inactivation of tumour suppressor proteins. Interestingly, some well-known tumour suppressor proteins have been described to combine key regulatory functions of signalling pathways with protection from CIN. In such circumstance, loss of a tumour suppressor protein activity deregulates both key cell signalling pathways as well as chromosomal stability mechanisms. These double functions are described for several tumour suppressors including adenomatous polyposis coli (APC), retinoblastoma (RB), the RE1-silencing transcription factor (REST), the tumour suppressor protein p53 (TP53) and the von Hippel-Lindau protein (pVHL) (table 1).
|
Table 1: Tumour suppressor proteins and their function in suppression of aneuploidy. |
|
Tumor suppressor
protein
|
Tumor suppressor
function
|
Aneuploidy suppressor
function
|
| APC |
Canonical Wnt signalling, involved in cytoplasmic destruction complex targeting beta-catenin for degradation [92]
Regulation of the cytoskeleton and cell migration [93] |
Promotion of stable kinetochore-microtubule attachments to suppress W-CIN [91]
64 |
| TP53 |
Sensor for DNA damage and activator of repair pathways [94]
Initiation of apoptosis and senescence [95] |
Control of centrosome duplication [67]
96 |
| RB |
Control of G1-S transition via E2F inhibition
Chromatin remodeling and maintenance of a quiescent state [97] |
Suppression of W-CIN by transcriptional control of Mad2 via E2F [74]
71 |
| REST |
Suppression of PI(3)-Kinase signalling [78] |
Suppression of W-CIN by transcriptional repression of Mad2[81] |
| VHL |
Hypoxia response, involved in an E3-ligase complex targeting Hif-alpha transcription factors [98]
Regulation of extracellular matrix deposition and microtubule stability [83] |
Control of SAC by maintaining Mad2 protein levels to suppress W-CIN [90] |
Adenomatous polyposis coli protein (APC)
Mutations of the apcgene are a hallmark of familial adenomatous polyposis (FAP), where some of these polyps develop to malignant carcinomas. In addition apc mutations are also found in about 85% of sporadic colorectal cancers [57]. APC’s most attributed tumour-suppressor function is as a negative regulator of the canonical Wnt signaling pathway [58]. Furthermore actin and microtubule (MT) regulatory roles have been described [59], the latter has been also linked to CIN.
In this context, APC has been described as a MT plus-end interacting protein (+Tip) [60, 61] with a role in stabilizing astral MTs during mitosis. Furthermore localization to the kinetochore has also been reported [62, 63]. At kinetochores APC may influence mitosis in two ways: by regulating kinetochore MT stability and tethering to kinetochores as well as regulating the SAC, indeed reduced tension at the metaphase plate is observed after depletion of APC by RNAi in U2OS cells [64]. The opposite effect is observed in HeLa cells [91]. Despite these cell-line specific discrepancies, in both cases the observed kinetochore-phenotypes result in a defect in maintaining an accurate chromosome number. However, the observed aneuploidy could also be a result of defects in the SAC due to APC loss. APC interacts with Bub1 and BubR1 and is consequently phosphorylated in vitro by these Bub proteins [63, 65]. In addition reduced localisation of Bub1 and BubR1 to kinetochores are observed, at least in U2OS cells, as a result of APC depletion [64]. Despite these discrepancies, a current view ascribes a defect in mediating MT-kinetochore attachment as a primary cause of the observed CIN in APC-negative cells.
Tumour suppressor protein p53 (TP53)
The tumour suppressor protein 53 (TP53), also called “the guardian of the genome”, is a member of the p53 transcription factor family. The TP53 protein is an integrator of various cellular stress signals, like DNA damage and oxidative stress. TP53 responds to this stresses by transcribing genes that lead to a cell cycle arrest or, if the damage due to the stress is irreparable, to apoptotis [66]. Besides these extensively studied and reviewed functions, TP53 is also implicated in the control of the mitotic checkpoint in several ways. There is evidence that TP53 is involved in centrosome duplication, since mouse embryonic fibro-blasts lacking p53 have an increased number of functional centrosomes resulting in merotelic kinetochore attachments and segregation errors [67] (fig. 1B). Furthermore it has been reported that the mitotic checkpoint protein BubR1 is a transcriptional target of p53 [68]. BubR1 expression is indeed considerably low in cells lacking p53 expression and the checkpoint is therefore compromised. Whether these two processes in the mitotic checkpoint affected by loss of p53 function contribute to the TP53 tumour suppressive mechanism is however still unclear. Nevertheless, these molecular linkages linking p53 function to the regulation of mitotic events warrant further exploration.
Retinoblastoma (RB)
Mutations in the Retinoblastoma (Rb) tumour suppressor gene have been originally discovered in a rare childhood cancer called retinoblastoma [69]. Rb mutations are found in a wide variety of cancers mainly in the lung, breast and eye [5]. In cervical and other squamous cell carcinomas the retinoblastoma protein (pRB) is frequently inactivated by expression of the human papillomavirus (HPV) E7 protein. pRB guards the cell from replicating damaged DNA and therefore prevents the amplification and integration of mutations into the genome by blocking cell cycle progression from the first gap phase (G1) into the DNA synthesis phase (S). RB is assigned a potent role as a regulator of the G1/S transition. This tumour suppressive function is exerted mainly by the binding to and sequestration of the E2F family transcription factors by pRB [70]. Reversion of this interaction occurs when cells encounter persistent mitogenic stimuli sufficient for the activation of cyclin dependent kinases (CDK) that phosphorylate pRB in the nucleus. This reliefs E2Fs and therefore cancels pRBs growth inhibitory function and facilitates S phase entry.
To build the mitotic spindle after S-phase, the centrosome has to be duplicated in a coordinated manner. The RB-E2F pathway contributes to this coordination and inactivation of this pathway jeopardizes the coordination between DNA replication and centrosome duplication potentially contributing to genomic instability [71]. Furthermore, cells with inactivated Rb that have been reversibly blocked in mitosis and subsequently released from this block accumulate an abnormal number of chromosomes [72]. Additional evidence that pRB has an important function besides the G1/S transition control comes from studies in mouse embryonic stem cells. Mouse embryonic stem cells are largely devoid of a G1/S checkpoint. Interestingly, deletion of both Rb alleles in mouse embryonic stem cells increases chromosomal alterations [73]. The mitotic checkpoint gene Mad2 is a direct E2F transcriptional target [74] and is aberrantly expressed in cells with defects in the RB pathway. Indeed, Mad2 overexpresison correlates with heightened E2F activity in tumours as well as in several cell lines resulting in mitotic defects and aneuploidy as a consequence. Another mitotic role of pRB that is independent of pRB’s ability to interact with E2F has been proposed in the control of condensin II complex formation to regulate appropriate pericentric heterochromatin formation and chromosome condensation during G2/M [75–77]. Although the phenotype is subtle and still allows the cells to divide, failure of this function is thought to lead to CIN.
RE1-silencing transcription factor (REST)
The regulation of Mad2 checkpoint protein levels seems also to play a critical role in the tumour suppressor function of the repressor-element-1-silencing transcription factor (REST). Initially a tumour suppressor function of REST has been observed in a genomic screen aimed to identify novel potential tumour suppressor proteins. It has been shown that REST protects primary cell transformation by suppressing the PI(3)-kinase pathway [78]. REST is a transcriptional repressor and regulates gene expression via binding to a 21–23 base pair repressor element in the promoters of relevant target genes. Depending on the cellular context, REST can act either oncogenic or tumour-suppressive. In its oncogenic context, high levels of truncated forms of REST are found in small-cell lung cancer and neuroblastoma [79, 80], whereas a frame-shift mutant of REST is found in colon cancer that is able to transform primary cells [78]. Amongst the many repressed gene targets of REST, there is also Mad2 [81]. A regulatory network that involves the proteasomal degradation of REST by the E3 ubiquitin ligase complex Skp1-Cul1-F-βTRCP [81, 82] during the G2-phase of the cell cycle reliefs the transcriptional repression of Mad2 by REST and allows a controlled activation of the mitotic checkpoint. Expression of oncogenic truncation variants of REST that escape the E3 ubiquitin ligase regulation, suppress Mad2 protein levels, resulting in a defective spindle-checkpoint and aneuploidy [81].
Von Hippel-Lindau protein (pVHL)
Maintaining appropriate Mad2 protein levels and suppression of aneuploidy is also a critical function of the von Hippel-Lindau (VHL) tumour suppressor protein. Mutations of the VHL tumour suppressor gene occur in a variety of inherited and sporadic tumours, including clear cell renal cell carcinoma. A multitude of tumour suppressor functions have been attributed to the VHL gene product [83], pVHL, amongst these are the degradation of the hypoxia inducible factors (HIF)-alpha [84], stabilization of microtubules [85, 86], primary cilium maintenance [87, 88] and regulation of extracellular matrix deposition [89]. Analysis of the cell cycle in cells devoid of pVHL revealed two unexpected defects: rotating mitotic spindles and a weakened mitotic checkpoint due to lower protein levels of Mad2, which resulted in premature mitotic slippage and whole-chromosome aneuploidy in primary cells [90]. Reconstitution of pVHL in VHL-negative kidney cancer cells re-established Mad2 protein levels, prevented mitotic slippage and suppressed aneuploidy in vitro. Evidence, that this regulatory function of the mitotic checkpoint via Mad2 is critical for suppression of aneuploidy is supported by the observation that inactivation of VHL in human clear cell renal cell carcinoma correlates with a reduction of Mad2 protein levels and that this is associated with enhanced aneuploidy and tumour-grade [90]. These results link functional loss of pVHL to reduced Mad2 protein and the development of aneuploidy. Open issues are now to identify the relevant mechanism of pVHL control of Mad2 abundance and to determine whether this regulatory mechanism mediated by pVHL is critical for its tumour suppression function.
Conclusion
Defective mitotic checkpoint signalling that is compatible with cell life appears to underlie the development of aneuploidy, a potential significant contributor of tumour cell evolution. Several tumour suppressor proteins share the ability to suppress aneuploidy, albeit through distinct mechanisms. It is therefore tempting to propose that the shared additional function of these tumour-suppressor proteins in protecting cells from CIN might be a significant additional fuel to drive tumour progression.
W-CIN is mainly driven by a defective mitotic checkpoint or a defective centrosome duplication cycle and the level of deregulation often is critical for a cell to be transformed and hyper-proliferative or for cell death. Knowledge about the nature of such checkpoint deregulation signatures in specific tumour-types elicited by e.g., loss of tumour-suppressor proteins as well as other characteristic genetic alterations might provide specific druggable targets to tip the balance in the tumour cells towards cell death, especially since many mitotic drugs already exist.
We thank all members of our laboratories for helpful discussions. And apologise to all authors whose significant contributions could not be cited due to space limitations.
Correspondence to:
Professor Wilhelm Krek
Institute of Cell Biology
HPM, F 42
Schafmattstrasse 18
CH-8093 Zürich
Switzerland
wilhelm.krek@cell.biol.ethz.ch
References
1 Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70.
2 Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell. 2008;13:472–82.
3 Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–37.
4 Hahn WC, Weinberg RA. Modelling the molecular circuitry of cancer. Nat Rev Cancer. 2002;2:331–41.
5 COSMIC. (2009) Catalog of somatic mutations in cancer. http://www.sanger.ac.uk/genetics/CGP/cosmic/.
6 Boveri T. Zur Frage der Entstehung maligner Tumoren. Gustav Fischer, Jena, 1914.
7 Mitelman F, Johansson B, Mertens F. (2010) Mitelman Database of Chromosome Aberrations in Cancer. http://cgap.nci.nih.gov/Chromosomes/Mitelman.
8 Duesberg P, Li R. Multistep carcinogenesis: a chain reaction of aneuploidizations. Cell Cycle. 2003;2:202–10.
9 Zimonjic D, Brooks MW, Popescu N, Weinberg RA, Hahn WC. Derivation of human tumor cells in vitro without widespread genomic instability. Cancer Res. 2001;61:8838–44.
10 Weaver BA, Silk AD, Montagna C, Verdier-Pinard P, Cleveland DW. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell. 2007;11:25–36.
11 Gasparini P, Sozzi G, Pierotti MA. The role of chromosomal alterations in human cancer development. J Cell Biochem. 2007;102:320–31.
12 Rowley JD. Identificaton of a translocation with quinacrine fluorescence in a patient with acute leukemia. Ann Genet. 1973;16:109–12.
13 Sandberg AA, Meloni-Ehrig AM. Cytogenetics and genetics of human cancer: methods and accomplishments. Cancer Genet Cytogenet. 2010;203:102–26.
14 Lingle WL, Lukasiewicz K, Salisbury JL. Deregulation of the centrosome cycle and the origin of chromosomal instability in cancer. Adv Exp Med Biol. 2005;570:393–421.
15 Cimini D, Wan X, Hirel CB, Salmon ED. Aurora kinase promotes turnover of kinetochore microtubules to reduce chromosome segregation errors. Curr Biol. 2006;16:1711–8.
16 Knowlton AL, Lan W, Stukenberg PT. Aurora B is enriched at merotelic attachment sites, where it regulates MCAK. Curr Biol. 2006;16:1705–10.
17 Cimini D. Detection and correction of merotelic kinetochore orientation by Aurora B and its partners. Cell Cycle. 2007;6:1558–64.
18 Musacchio A, Salmon ED. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol. 2007;8:379–93.
19 Cheeseman IM, Desai A. Molecular architecture of the kinetochore-microtubule interface. Nat Rev Mol Cell Biol. 2008;9:33–46.
20 Rieder CL, Cole RW, Khodjakov A, Sluder G. The checkpoint delaying anaphase in response to chromosome monoorientation is mediated by an inhibitory signal produced by unattached kinetochores. J Cell Biol. 1995;130:941–8.
21 Sironi L, Mapelli M, Knapp S, De Antoni A, Jeang KT, Musacchio A. Crystal structure of the tetrameric Mad1-Mad2 core complex: implications of a “safety belt” binding mechanism for the spindle checkpoint. Embo J. 2002;21:2496–506.
22 Chan GK, Jablonski SA, Sudakin V, Hittle JC, Yen TJ. Human BUBR1 is a mitotic checkpoint kinase that monitors CENP-E functions at kinetochores and binds the cyclosome/APC. J Cell Biol. 1999;146:941–54.
23 Yao X, Abrieu A, Zheng Y, Sullivan KF, Cleveland DW. CENP-E forms a link between attachment of spindle microtubules to kinetochores and the mitotic checkpoint. Nat Cell Biol. 2000;2:484–91.
24 Johnson VL, Scott MI, Holt SV, Hussein D, Taylor SS. Bub1 is required for kinetochore localization of BubR1, Cenp-E, Cenp-F and Mad2, and chromosome congression. J Cell Sci. 2004;117:1577–89.
25 Meraldi P, Sorger PK. A dual role for Bub1 in the spindle checkpoint and chromosome congression. Embo J. 2005;24:1621–33.
26 De Antoni A, Pearson CG, Cimini D, Canman JC, Sala V, Nezi L, et al. The Mad1/Mad2 complex as a template for Mad2 activation in the spindle assembly checkpoint. Curr Biol. 2005;15:214–25.
27 Mapelli M, Massimiliano L, Santaguida S, Musacchio A. The Mad2 conformational dimer: structure and implications for the spindle assembly checkpoint. Cell. 2007;131:730–43.
28 Luo X, Fang G, Coldiron M, Lin Y, Yu H, Kirschner MW, Wagner G. Structure of the Mad2 spindle assembly checkpoint protein and its interaction with Cdc20. Nat Struct Biol. 2000;7:224–9.
29 Kim Y, Heuser JE, Waterman CM, Cleveland DW. CENP-E combines a slow, processive motor and a flexible coiled coil to produce an essential motile kinetochore tether. J Cell Biol. 2008;181:411–9.
30 Mao Y, Abrieu A, Cleveland DW. Activating and silencing the mitotic checkpoint through CENP-E-dependent activation/inactivation of BubR1. Cell. 2003;114:87–98.
31 Howell BJ, McEwen BF, Canman JC, Hoffman DB, Farrar EM, Rieder CL, Salmon ED. Cytoplasmic dynein/dynactin drives kinetochore protein transport to the spindle poles and has a role in mitotic spindle checkpoint inactivation. J Cell Biol. 2001;155:1159–72.
32 Wojcik E, Basto R, Serr M, Scaerou F, Karess R, Hays T. Kinetochore dynein: its dynamics and role in the transport of the Rough deal checkpoint protein. Nat Cell Biol. 2001;3;1001–7.
33 Michel LS, Liberal V, Chatterjee A, Kirchwegger R, Pasche B, Gerald W, et al. MAD2 haplo-insufficiency causes premature anaphase and chromosome instability in mammalian cells. Nature. 2001;409:355–9.
34 Sotillo R, Hernando E, Diaz-Rodriguez E, Teruya-Feldstein J, Cordon-Cardo C, Lowe SW, Benezra R. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell. 2007;11:9–23.
35 Cahill DP, Lengauer C, Yu J, Riggins GJ, Willson JK, Markowitz SD, et al. Mutations of mitotic checkpoint genes in human cancers. Nature. 1998;392:300–3.
36 Weaver BA, Cleveland DW. Does aneuploidy cause cancer? Curr Opin Cell Biol. 2006;18:658–67.
37 Dobles M, Liberal V, Scott ML, Benezra R, Sorger PK. Chromosome missegregation and apoptosis in mice lacking the mitotic checkpoint protein Mad2. Cell. 2000;101:635–45.
38 Iwanaga Y, Chi YH, Miyazato A, Sheleg S, Haller K, Peloponese JM Jr, et al. Heterozygous deletion of mitotic arrest-deficient protein 1 (MAD1) increases the incidence of tumors in mice. Cancer Res. 2007;67:160–6.
39 Jeganathan K, Malureanu L, Baker DJ, Abraham SC, van Deursen JM. Bub1 mediates cell death in response to chromosome missegregation and acts to suppress spontaneous tumorigenesis. J Cell Biol. 2007;179:255–67.
40 Kalitsis P, Earle E, Fowler KJ, Choo KH. Bub3 gene disruption in mice reveals essential mitotic spindle checkpoint function during early embryogenesis. Genes Dev. 2000;1:2277–82.
41 Wang Q, Liu T, Fang Y, Xie S, Huang X, Mahmood R, et al. BUBR1 deficiency results in abnormal megakaryopoiesis. Blood. 2004;103:1278–85.
42 Weaver BA, Bonday ZQ, Putkey FR, Kops GJ, Silk AD, Cleveland DW. Centromere-associated protein-E is essential for the mammalian mitotic checkpoint to prevent aneuploidy due to single chromosome loss. J Cell Biol. 2003;162:551–63.
43 Perez de Castro I, de Carcer G, Malumbres M. A census of mitotic cancer genes: new insights into tumor cell biology and cancer therapy. Carcinogenesis. 2007;28:899–912.
44 Kalitsis P, Fowler KJ, Griffiths B, Earle E, Chow CW, Jamsen K, Choo KH. Increased chromosome instability but not cancer predisposition in haploinsufficient Bub3 mice. Genes Chromosomes Cancer. 2005;44:29–36.
45 Baker DJ, Jeganathan KB, Cameron JD, Thompson M, Juneja S, Kopecka A, et al. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat Genet. 2004;36:744–9.
46 Dai W, Wang Q, Liu T, Swamy M, Fang Y, Xie S, Mahmood R, et al. Slippage of mitotic arrest and enhanced tumor development in mice with BubR1 haploinsufficiency. Cancer Res. 2004;64:440–5.
47 Babu JR, Jeganathan KB, Baker DJ, Wu X, Kang-Decker N, van Deursen JM. Rae1 is an essential mitotic checkpoint regulator that cooperates with Bub3 to prevent chromosome missegregation. J Cell Biol. 2003;160:341–53.
48 Foijer F, Draviam VM, Sorger PK. Studying chromosome instability in the mouse. Biochim Biophys Acta. 2008;1786:73–82.
49 Schvartzman JM, Sotillo R, Benezra R. Mitotic chromosomal instability and cancer: mouse modelling of the human disease. Nat Rev Cancer. 10:102–15.
50 Segal DJ, McCoy EE. Studies on Down’s syndrome in tissue culture. I. Growth rates and protein contents of fibroblast cultures. J Cell Physiol. 1974;83:85–90.
51 Torres EM, Sokolsky T, Tucker CM, Chan LY, Boselli M, Dunham MJ, Amon A. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science. 2007;317:916–24.
52 Williams BR, Prabhu VR, Hunter KE, Glazier CM, Whittaker CA, Housman DE, Amon A. Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science. 2008;322:703–9.
53 Williams BR, Amon A. Aneuploidy: cancer’s fatal flaw? Cancer Res. 2009;69:5289–91.
54 Nicholson JM, Duesberg P. On the karyotypic origin and evolution of cancer cells. Cancer Genet Cytogenet. 2009;194:96–110.
55 Torres EM, Dephoure N, Panneerselvam A, Tucker CM, Whittaker CA, Gygi SP, et al. Identification of aneuploidy-tolerating mutations. Cell. 2010;143:71–83.
56 Sotillo R, Schvartzman JM, Socci ND, Benezra R. Mad2-induced chromosome instability leads to lung tumour relapse after oncogene withdrawal. Nature.
57 Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–70.
58 Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–80.
59 Nathke IS. The adenomatous polyposis coli protein: the Achilles heel of the gut epithelium. Annu Rev Cell Dev Biol. 2004;20:337–66.
60 Mimori-Kiyosue Y, Shiina N, Tsukita S. Adenomatous polyposis coli (APC) protein moves along microtubules and concentrates at their growing ends in epithelial cells. J Cell Biol. 2000;148:505–18.
61 Nathke IS, Adams CL, Polakis P, Sellin JH, Nelson WJ. The adenomatous polyposis coli tumor suppressor protein localizes to plasma membrane sites involved in active cell migration. J Cell Biol. 1996;134:165–79.
62 Fodde R, Kuipers J, Rosenberg C, Smits R, Kielman M, Gaspar C, et al. Mutations in the APC tumour suppressor gene cause chromosomal instability. Nat Cell Biol. 2001;3:433–8.
63 Kaplan KB, Burds AA, Swedlow JR, Bekir SS, Sorger PK, Nathke IS. A role for the Adenomatous Polyposis Coli protein in chromosome segregation. Nat Cell Biol. 2001;3:429–32.
64 Dikovskaya D, Schiffmann D, Newton IP, Oakley A, Kroboth K, Sansom O, et al. Loss of APC induces polyploidy as a result of a combination of defects in mitosis and apoptosis. J Cell Biol. 2007;176:183–95.
65 Zhang J, Ahmad S, Mao Y. BubR1 and APC/EB1 cooperate to maintain metaphase chromosome alignment. J Cell Biol. 2007;178:773–84.
66 Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability – an evolving hallmark of cancer. Nat Rev Mol Cell Biol. 2010;11:220–8.
67 Fukasawa K, Choi T, Kuriyama R, Rulong S, Vande Woude GF. Abnormal centrosome amplification in the absence of p53. Science. 1996;271:1744–7.
68 Oikawa T, Okuda M, Ma Z, Goorha R, Tsujimoto H, Inokuma H, Fukasawa K. Transcriptional control of BubR1 by p53 and suppression of centrosome amplification by BubR1. Mol Cell Biol. 2005;25:4046–61.
69 Sabado Alvarez C. Molecular biology of retinoblastoma. Clin Transl Oncol. 2008;10:389–94.
70 Chen HZ, Tsai SY, Leone G. Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nat Rev Cancer. 2009;9:785–97.
71 Meraldi P, Lukas J, Fry AM, Bartek J, Nigg EA. Centrosome duplication in mammalian somatic cells requires E2F and Cdk2-cyclin A. Nat Cell Biol. 1999;1:88–93.
72 Lentini L, Pipitone L, Di Leonardo A. Functional inactivation of pRB results in aneuploid mammalian cells after release from a mitotic block. Neoplasia. 2002;4:380–7.
73 Zheng L, Flesken-Nikitin A, Chen PL, Lee WH. Deficiency of Retinoblastoma gene in mouse embryonic stem cells leads to genetic instability. Cancer Res. 2002;62:2498–502.
74 Hernando E, Nahle Z, Juan G, Diaz-Rodriguez E, Alaminos M, Hemann M, et al. Rb inactivation promotes genomic instability by uncoupling cell cycle progression from mitotic control. Nature. 2004;430:797–802.
75 Coschi CH, Martens AL, Ritchie K, Francis SM, Chakrabarti S, Berube NG, Dick FA. Mitotic chromosome condensation mediated by the retinoblastoma protein is tumor-suppressive. Genes Dev. 2010;24:1351–63.
76 Manning AL, Longworth MS, Dyson NJ. Loss of pRB causes centromere dysfunction and chromosomal instability. Genes Dev. 2010;24:1364–76.
77 van Harn T, Foijer F, van Vugt M, Banerjee R, Yang F, Oostra A, et al. Loss of Rb proteins causes genomic instability in the absence of mitogenic signaling. Genes Dev. 2010;24:1377–88.
78 Westbrook TF, Martin ES, Schlabach MR, Leng Y, Liang AC, Feng B, et al. A genetic screen for candidate tumor suppressors identifies REST. Cell. 2005;121:837–48.
79 Coulson JM, Edgson JL, Woll PJ, Quinn JP. A splice variant of the neuron-restrictive silencer factor repressor is expressed in small cell lung cancer: a potential role in derepression of neuroendocrine genes and a useful clinical marker. Cancer Res. 2000;60:1840–4.
80 Palm K, Metsis M, Timmusk T. Neuron-specific splicing of zinc finger transcription factor REST/NRSF/XBR is frequent in neuroblastomas and conserved in human, mouse and rat. Brain Res Mol Brain Res. 1999;72:30–9.
81 Guardavaccaro D, Frescas D, Dorrello NV, Peschiaroli A, Multani AS, Cardozo T, et al. Control of chromosome stability by the beta-TrCP-REST-Mad2 axis. Nature. 2008;452:365–9.
82 Westbrook TF, Hu G, Ang XL, Mulligan P, Pavlova NN, Liang A, et al. SCFbeta-TRCP controls oncogenic transformation and neural differentiation through REST degradation. Nature. 2008;452:370–4.
83 Frew IJ, Krek W. pVHL: a multipurpose adaptor protein. Sci Signal, 1, pe30. (2008).
84 Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–5.
85 Hergovich A, Lisztwan J, Barry R, Ballschmieter P, Krek W. Regulation of microtubule stability by the von Hippel-Lindau tumour suppressor protein pVHL. Nat Cell Biol. 2003;5:64–70.
86 Thoma CR, Matov A, Gutbrodt KL, Hoerner CR, Smole Z, Krek W, Danuser G. Quantitative image analysis identifies pVHL as a key regulator of microtubule dynamic instability. J Cell Biol. 2010;190:991–1003.
87 Schermer B, Ghenoiu C, Bartram M, Muller RU, Kotsis F, Hohne M, et al. The von Hippel-Lindau tumor suppressor protein controls ciliogenesis by orienting microtubule growth. J Cell Biol. 2006;175:547–54.
88 Thoma CR, Frew IJ, Hoerner CR, Montani M, Moch H, Krek W. pVHL and GSK3beta are components of a primary cilium-maintenance signalling network. Nat Cell Biol. 2007;9:588–95.
89 Ohh M, Yauch RL, Lonergan KM, Whaley JM, Stemmer-Rachamimov AO, Louis DN, et al. The von Hippel-Lindau tumor suppressor protein is required for proper assembly of an extracellular fibronectin matrix. Mol Cell. 1998;1:959–68.
90 Thoma CR, Toso A, Gutbrodt KL, Reggi SP, Frew IJ, Schraml P, et al. VHL loss causes spindle misorientation and chromosome instability. Nat Cell Biol. 2009;11:994–1001.
91 Draviam VM, Shapiro I, Aldridge B, Sorger PK. Misorientation and reduced stretching of aligned sister kinetochores promote chromosome missegregation in EB1- or APC-depleted cells. Embo J. 2006;25:2814–27.
92 Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer. 2008;8:387-98.
93 Nathke I. Relationship between the role of the adenomatous polyposis coli protein in colon cancer and its contribution to cytoskeletal regulation. Biochem Soc Trans. 2005;33:694-7.
94 Meek DW. Tumour suppression by p53: a role for the DNA damage response? Nat Rev Cancer. 2009;9:714-23.
95 Zuckerman V. Wolyniec K, Sionov RV, Haupt S, Haupt Y. Tumour suppression by p53: the importance of apoptosis and cellular senescence. J Pathol. 2009;219:3-15.
96 Thompson SL, Compton DA. Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J Cell Biol. 2010;188;369-81.
97 Burkhart DL, Sage J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer. 2008;8:671-82.
98 Kaelin WG, Jr. The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer. Nat Rev Cancer. 2008;8:865-73.