Inhibition of NF-κB and AP-1 by dimethylfumarate correlates with down-regulated IL-6 secretion and proliferation in human lung fibroblasts
DOI: https://doi.org/10.4414/smw.2010.13132
Summary
QUESTION UNDER STUDY/PRINCIPLES: Dimethylfumarate (DMF) had been reported to reduce asthma symptoms and to improve the quality of life of asthma patients. Therefore, we assessed the anti-inflammatory and anti-remodeling effect of DMF on isolated lung fibroblasts, which are relevant to inflammatory lung diseases. We determined the effect of DMF on platelet derived growth factor (PDGF)-BB induced proliferation, as well as on PDGF-BB and tumor necrosis factor (TNF)-α induced interleukin (IL)-6 secretion and on activation of activated protein (AP)-1 and nuclear factor kappaB (NF-ĸB).
METHODS: Confluent lung fibroblasts were incubated with DMF (0.1–100 μM) 1 hour before stimulation with PDGF-BB or TNF-α (both 10 ng/ml). IL-6 secretion was measured by ELISA. NF-ĸB and AP-1 activation was determined by immuno-blotting and EMSA. Cell proliferation was assessed by [3H]-thymidine incorporation in subconfluent fibroblasts.
RESULTS: PDGF-BB but not TNF-α induced fibroblast proliferation. This was dose dependently reduced by DMF in a concentration range of 10–100 μM. PDGF-BB and TNF-α induced IL-6 secretion by lung fibroblasts and this was inhibited by DMF in a dose-dependent manner. However, PDGF-BB induced IL-6 secretion did not correlate with NF-ĸB activity, while TNF-α did. DMF inhibited the TNF-α induced NF-ĸB–DNA binding, but had neither an inhibitory effect on NF-ĸB nuclear entry nor on the degradation of IκB-α. PDGF-BB and TNF-α activated AP-1, which was also inhibited by DMF.
CONCLUSIONS: Our data suggest that DMF down-regulates TNF-α-induced IL-6 secretion and proliferation by inhibiting NF-ĸB and AP-1 activity, indicating its potential beneficial use for the treatment of inflammatory lung diseases.
Introduction
Fumaric acid esters (FAE) are used to treat psoriasis and in Germany, a mixture of dimethylfumarate (DMF) is registered for the management of severe psoriasis [1]. The clinical efficacy and safety of an oral FAE formulation (BG00012), which contains only DMF is documented in multiple sclerosis [2]. Occasional reports indicated that DMF reduced symptoms and improved the quality of life of asthma patients.
Airway inflammation and remodeling in inflammatory lung diseases such as asthma and chronic obstructive pulmonary disease (COPD) is a multi-level process involving T-cells, neutrophils and eosinophils [3]. In addition to these classical inflammatory cells, activated lung fibroblasts and airway smooth muscle cells contribute to the pathology of inflammatory lung diseases by secreting a wide range of pro-inflammatory factors [4–6]. Today, inhaled glucocorticoids (GC) and long acting β2-agonists are the most frequently applied drugs for inflammatory lung diseases [7]. However, in chronic inflammatory lung diseases, airway inflammation and remodeling are not well controlled by any of the available therapeutic strategies [8]. Therefore, new therapeutic alternatives are needed to control airway inflammation in inflammatory lung diseases.
Interleukin (IL)-6 is considered to contribute to the severity of inflammatory lung diseases [9]. The IL-6 promoter is regulated by the activator protein (AP)-1, and nuclear factor (NF)-ĸB [10]. Both, NF-ĸB and AP-1, are considered as new targets for pharmacological approaches in asthma therapy [11]. In human keratinocytes DMF inhibited the activation of NF-ĸB and subsequently the expression of IL-8 and IL-20 [12]. In human T-cells, DMF reduced the binding of active NF-ĸB to its corresponding DNA consensus sequence [13]. The nuclear accumulation of NF-ĸB p50 and the subsequent expression of ICAM-1 were inhibited by DMF in human dermal fibroblasts [14]. Furthermore, DMF inhibited the TNF-α induced nuclear entry of NF-ĸB p65 and p50 thereby reducing the expression of tissue factor mRNA and protein by human endothelial cells [15]. In primary human airway smooth muscle cells DMF inhibited the NF-ĸB signaling cascade on multiple levels thereby down-regulating the secretion of asthma relevant eotaxin, RANTES and IL-6. Interestingly, our data indicated that DMF directly interacts with NF-κB thereby reducing its binding capacity to the corresponding DNA consensus sequence [6].
Based on histo-pathological studies fibroblasts play a role in airway remodeling in chronic inflammatory diseases and represent the major producer of extra-cellular matrix which increases airway wall thickness [16–18]. Airway remodeling often occurs before any sign of inflammation can be detected and may trigger inflammation [17, 19, 20]. By secreting thymus- and activation-regulated chemokine (TARC) fibroblasts recruit Th2 cells to the inflamed section of the lung [21].
The aim of this study was to determine the effect of DMF on PDGF-BB and TNF-α induced NF-ĸB and AP-1 activation, as well as on IL-6 secretion and fibroblast proliferation.
Methods
This study design was approved by the local ethics committee (University Hospital Basel, Switzerland) and written consent was obtained from all participants.
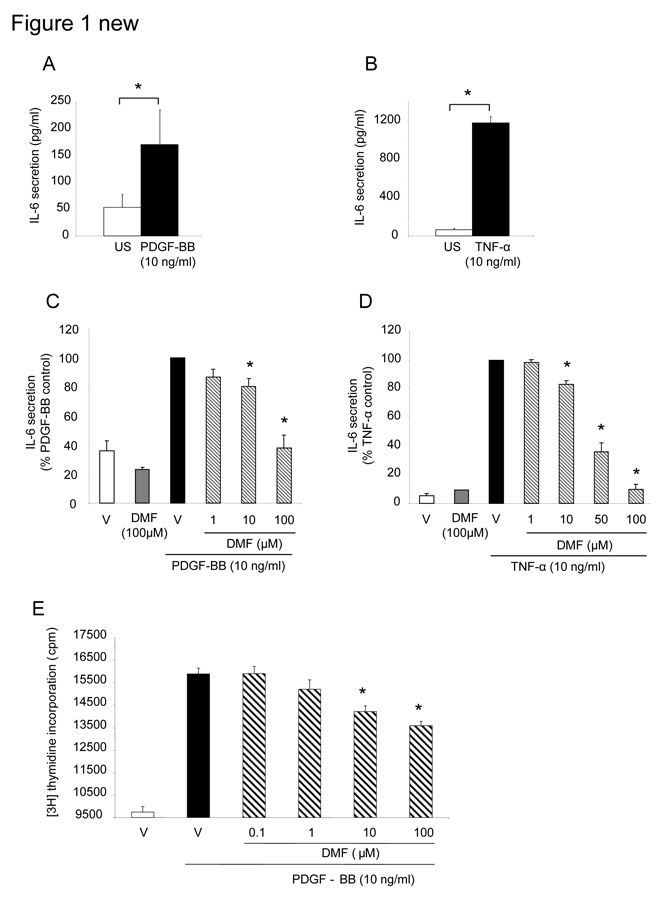
Figure 1
(A) IL-6 secretion induced by PDGF-BB (10 ng/ml) in human lung fibroblasts (n = 7). (*indicates p <0.05 student paired t-test). (B) TNF-α (10 ng/ml) stimulated IL-6 secretion in lung fibroblasts (n = 4) within 24h (* indicates p <0.0 5 student paired t-test). (C) DMF (1–100 µM) dose-dependently inhibited PDGF-BB or (D) TNF-α induced IL-6 secretion by lung fibroblasts (n = 5). The data is presented as mean ± SEM (* indicates p <0.05, one-way ANOVA). (E) DMF also inhibited PDGF-BB induced fibroblast proliferation. Data represents mean ± SEM (* indicates p <0.01, paired student’s t-test, two tailed).
Isolation of primary human lung fibroblasts: Fibroblasts were established from disease free lung parenchyma tissue (as defined by a pathologist) which was obtained from therapeutically resected lung tissue. Lung resections were performed to remove lung metastases of primary tumors in other organs. Tissue pieces (1–2 mm3) were placed into 25 cm2 flasks containing 1 ml of growth medium (RPMI1640, 8 mM stabilized L-glutamine, 20 mM Hepes, 5% fetal calf serum, 1% MEM vitamin mix; all Gibco/BRL, Switzerland) and were kept at standard cell culture conditions (37 °C, 100% humidity, 5% CO2, 95% air). All experiments were performed with fibroblasts between passages 3 to 6. Cells were serum deprived for 24 hours prior drug treatment and cytokine stimulation to prevent activation of various pro-inflammatory and pro-proliferative pathways by serum itself.
Drug preparation:DMF (Sigma, Buchs, Switzerland) was dissolved in dimethysulfoxide (DMSO; Sigma) and was further diluted in serum free medium.
IL-6 secretion by primary human lung cells: Confluent fibroblasts were serum starved for 24 hours before any treatment. DMF (0.1–100 μM) was added 1 hour prior to PDGF-BB or TNF-α stimulation (R&D Systems, Minneapolis, USA). After 24 hours cell culture medium samples were collected and IL-6 protein level was measured by enzyme-linked immunosorbent assay (R&D Systems).
Cell viability lactate dehydrogenase (LDH) membrane integrity assay:Fibroblasts were pre-treated with DMF (0.1–100 µM) for 1 hour and then stimulated with either 10 ng/ml TNF-α or PDGF-BB. After 24 hours the LDH membrane integrity assay was performed according to the manufactures protocol (CytoTox-OneTM, Promega, Madison, USA).
Isolation of nuclear and cytosolic proteins:NE-PER extraction kit (Pierce/Thermo Fisher Sci, Rockford, USA) was used to isolate nuclear and cytosolic proteins as follows: trypsinized cells were collected (3,000 rpm, 3 min) and incubated on ice with CER I buffer (10 min), than CER II buffer was added (1 min). Thenuclei were centrifuged (13.000 rpm, 4 °C, 5 min), and the supernatantwas collected as cytosolic protein fraction. The nuclei pellet wasre-suspended in NER buffer (40 min, 4 oC), than the cell debris was removed (13,000 rpm, 4 °C, 10 min) and the supernatantwas collected as nuclear protein fraction.
Electrophoretic Mobility Shift Assay (EMSA): The functional activity of transcription factors and their inhibition by DMF was studied by DNA-EMSA. Oligonucleotides were [γ33P]-dATP labeled (3000 Ci/mmol; Amersham Pharmacia Biotech, Freiburg, Germany) by T4 polynucleotide kinase (New England Biolabs, Hilden, Germany). Equal amounts of protein (5 μg) in 20 μg bovine serum albumin (Sigma), 2 μg poly dIdC (Roche Molecular Biochemicals, Basel, Switzerland), 1 μl Mg2Cl (only for AP-1), 2 μl D+-buffer [20 mM Hepes, (pH 7.9), 20% glycerol, 100 mM KCl, 0.5 mM EDTA, 0.25% NP-40, 2 mM DTT, 0.1% PMSF], 4 μl F-buffer (20% Ficoll 400, 100 mM Hepes, 300 mM KCl, 10 mM DTT, and 0.1% PMSF), and 100,000 cpm (Cerenkov) of [γ33P]-labeled NF-ĸB or AP-1 binding oligonucleotide (Promega) were mixed and adjusted to 20 μl. For supershift assays NF-ĸB p65 antibody: (H286; p50 :C19; Santa Cruz Biotech., Santa Cruz, USA) was added to the binding reaction and a 100-fold excess of unlabeled oligonucleotide was added to confirm specific AP-1 or NF-ĸB-DNA binding. Samples were incubated (room temperature, 25 min). and were size fractionated by electrophoresis 0.5x TBE (45 mM Tris-HCl, 45 mM boric acid, and 1 mM EDTA; 200V, 2 hours) in a non-denaturing 4% or 6% polyacrylamide gel. After electrophoresis, the gel was dried (1 hour, 80°C) and exposed to x-ray film (24 hours).
Immuno-blotting: Proteins were size-fractionated by electrophoresis (100 mM HEPES, 100 mM Tris base, 1% SDS; 140V, 60 min) in a 4-20% gradient PAGE-gel (Pierce). Protein transfer onto nitrocellulose membranes was confirmed by Ponceau’s staining [6]. Membranes were blocked by 5% w/v non-fat dry milk in Tris-buffered saline 0.1% Tween 20 (1 hour, room temperature) and then incubated with one of the primary antibodies: [anti- NF-ĸB/p65 (C-20), anti- NF-ĸB/p50 (H-119), anti-IĸB-α (C21) (all Santa Cruz)]. Primary antibodies were detected by horseradish peroxidase-conjugated IgG secondary antibodies diluted 1:2000–1:40000 (anti-rabbit IgG #7074, anti-mouse IgG #7076; Cell Signaling Tech., Beverly, USA) and protein bands were visualized by enhanced chemiluminescence (Pierce/Thermo Fisher Sci).
Proliferation: The effect of DMF on PDGF-BB induced cell proliferation was determined by [3H]-thymidine incorporation. Fibroblasts (5000 cells/well in 96-well plates) were grown to 60% confluence and then serum-deprived for 24 hours. Cells were pretreated with DMF (0.1-100 μM) for 1 hour and then stimulated with PDGF-BB (10 ng/ml) for 24 hours. 2 μCi/ml [3H]-thymidine (Amersham) was added to the cells 3 hours before incorporated [3H]-thymidine was determined by liquid scintillation counting.
Results
PDGF-BB (10 ng/ml) caused a weak, but significant increased of IL-6 secretion within 24 hours (p <0.05; student paired t-test; fig. 1A). Stimulating fibroblasts for 24 hours with 10 ng/ml TNF-α strongly induced IL-6 secretion [22] (fig. 1B).
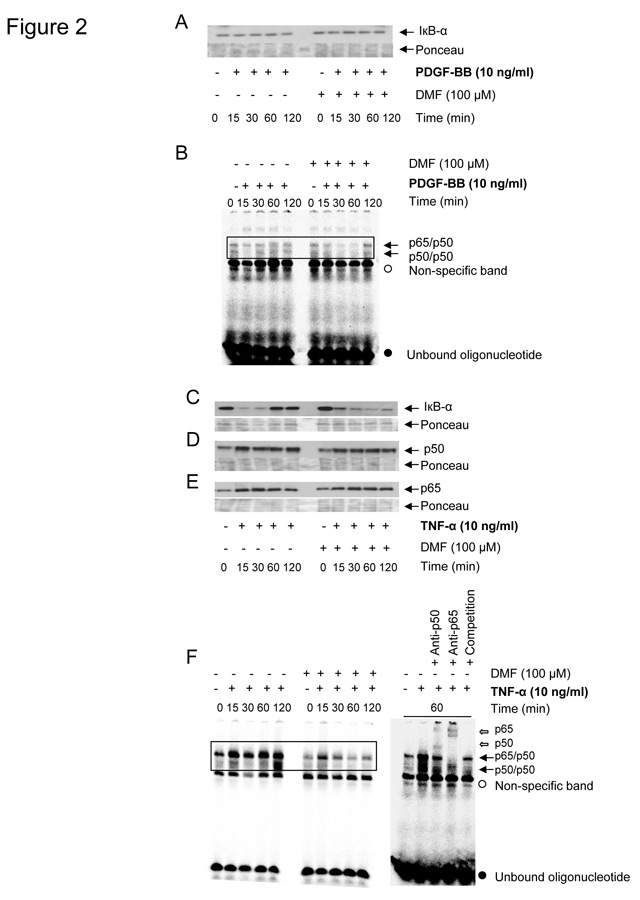
Figure 2
PDGF-BB (A, B) or TNF-α (C-F) specific effects of DMF (100 µM) on IĸB-α degradation, NF-ĸB p65/p50 nuclear entry and NF-ĸB to DNA binding in lung fibroblasts (n = 3). IĸB-α degradation in total cell lysates (A, C), and nuclear entry of NF-ĸB p65/p50 in nuclear cell lysates (D, E) are depicted as representative immuno-blots and similar results were obtained with two additional cell lines.
Pre-incubation (1 hour) with 10 µM DMF significantly decreased the secretion of PDGF-BB and TNF-α stimulated IL-6 by up to ~20% (p <0.0001 one-way ANOVA; fig. 1C, 1D). Pre-incubation with 50 µM DMF reduced TNF-α induced IL-6 secretion by 50% and further to background level at a concentration of 100 µM DMF (Figure 1D). Importantly, 100 µM DMF alone had no significant effect on IL-6 baseline levels (fig. 1C, 1D).
PDGF-BB (10 ng/ml) but not TNF-α at any concentration, strongly induced fibroblast proliferation within 24 hours by 155 ± 3.6%, and this effect was significantly decreased by DMF in a concentration range of 10 to 100 µM (p <0.05 unpaired student t-test; fig. 1E).
Lung fibroblasts that were pre-incubated with increasing concentrations of DMF (1–100 µM), or the drug vehicle DMSO (0.1%), or stimulated with PDGF-BB, or TNF-α (10 ng/ml) did not show any cytotoxic effect during 24 hours (data not shown).
PDGF-BB in the presence or absence of DMF 100 µM did neither induce IĸB-α degradation (fig. 2A), nor the binding of NF-ĸB to the DNA oligonucleotide (fig. 2B). In contrast, TNF-α stimulated IĸB-α degradation within 15–30 minutes, and IĸB-α was re-synthesized after 60–120 minutes (fig. 2C). Interestingly, pre-treatment with DMF extended the TNF-α induced degradation of IĸB-α for up to 120 minutes, while DMF alone did not affect IĸB-α stability (fig. 2C). TNF-α also induced (i) the nuclear entry of NF-ĸB p65/p50 (fig. 2D, 2E), (ii) DNA binding of NF-ĸB p65/p50, (iii) of NF-ĸB p50/p50 within 15 to 60 minutes (fig. 2F). Pre-incubation with DMF had no significant effect on TNF-α induced nuclear accumulation of NF-ĸB p65 or p50 (fig. 2D-2E), but inhibited the NF-ĸB binding to the DNA consensus sequence (fig. 2F).
PDGF-BB as well as TNF-α induced the activation and binding of AP-1 to a specific DNA oligonucleotide within 30–60 minutes (fig. 3A) and 30–120 minutes (fig. 3B), respectively. Both PDGF-BB and TNF-α induced AP-1 activation was inhibited when the fibroblasts had been pre-incubated for 1 hour with 100 µM DMF (fig. 3A, 3B). EMSA have been performed in a total of 3 fibroblast lines which yielded comparable results. The densitometric analysis of the AP-1 bands of the depicted representative EMSA are displayed in the small inserts in figure 3A and 3B. The bands representing the AP-1/DNA complex are not very defined, a phenomenon which we have observed in a previous publication [23] and which might be explained by the large number of different protein complexes of AP-1.
Discussion
In this study we provide evidence that DMF inhibits PDGF-BB and TNF-α induced IL-6 secretion as well as PDGF-BB induced proliferation in human primary lung fibroblasts. We further show that this anti-inflammatory and anti-proliferative effect of DMF correlated with a reduced activity of NF-ĸB and AP-1. We therefore propose that the inhibitory effect of DMF on NF-ĸB and AP-1 results in the reduction of IL-6 secretion and fibroblast proliferation.
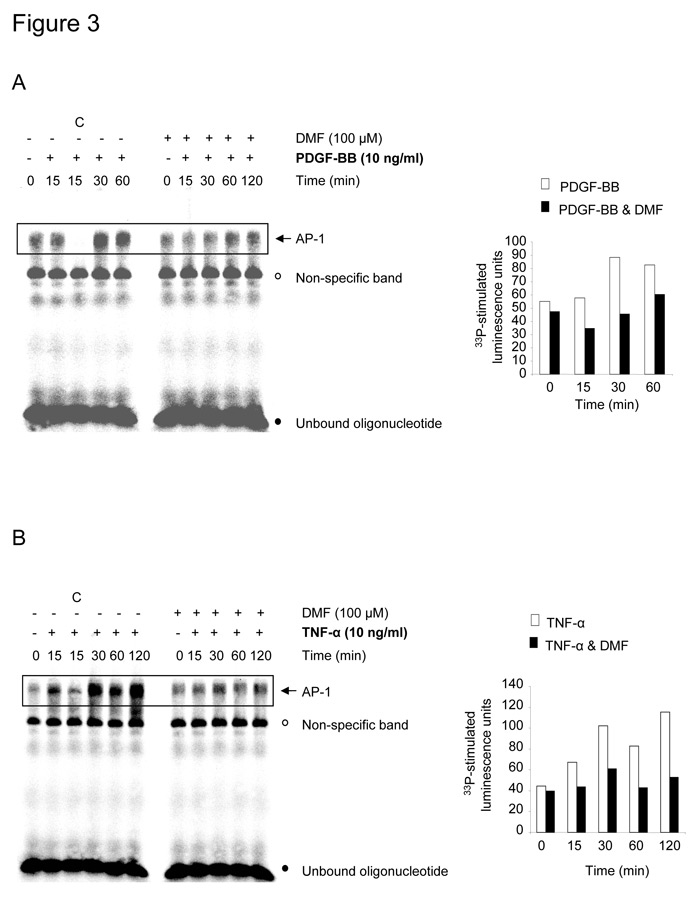
Figure 3
DMF (100 µM) inhibits PDGF-BB (A) or TNF-α (B) induced AP-1/DNA binding in lung fibroblasts (n = 3). The AP-1/DNA complex formation is shown as a representative EMSA which has been analyzed by densitometry, the result of which are shown as additional bar charts. Similar results were obtained in two additional experiments. Pre-incubation of extracts with a 100-fold excess of unlabeled oligonucleotides competed off the specific AP-1/DNA complexes (C: competition).
PDGF-BB stimulated remodeling and inflammation in the lung [22, 24], and stimulated proliferation and IL-6 synthesis via AP-1 and C/EBP-β in human lung mesenchymal cells [5, 25–28]. In rat airway smooth muscle cells PDGF-BB induced inflammation was mediated by NF-ĸB [29] and in human skin fibroblasts [20], but not in human vascular and airway smooth muscle cells [5, 31]. In our experiments, PDGF-BB neither degraded IĸB-α, nor stimulated NF-ĸB nuclear entry, nor NF-ĸB binding to its DNA consensus sequence, suggesting cell type or species specificity. Since the human IL-6 promoter contains an AP-1 binding site [10], this may explain our observation that DMF inhibited IL-6 secretion by PDGF-BB and TNF-α as well as proliferation, which also involves AP-1 [28].
TNF-α is elevated in the airways of patients with chronic inflammatory lung diseases and correlated with increased airways responsiveness [32]. Furthermore, TNF-α stimulates the secretion of pro-inflammatory cytokines by airway smooth muscle cells [7]. In our study in lung fibroblasts, TNF-α induced: (i) the degradation of IĸB-α, (ii) the nuclear entry of the NF-ĸB subunits p50 and p65, and (iii) NF-ĸB/DNA complex formation. DMF was reported to inhibit the nuclear entry and the binding of NF-ĸB to the DNA without inhibiting the degradation of IĸB in human airway smooth muscle [6]. Here we have shown that TNF-α induced NF-ĸB/DNA binding in lung fibroblasts was inhibited by DMF treatment. However, the nuclear entry of the NF-ĸB subunits p50 and p65 was not inhibited and IĸB-α re-synthesis was delayed by DMF after TNF-α stimulation. The delay in IĸB-α re-synthesis might be explained by the fact that the IĸB-α gene itself is regulated by NF-ĸB [33]. Thus, NF-ĸB inhibition may delay IĸB-α re-synthesis as it was previously described in mouse embryo fibroblasts [34]. This DMF cell type specificity might be important for its use as an anti-inflammatory and anti-remodeling drug in chronic inflammatory lung.
We have previously shown that DMF directly inhibits binding of active NF-ĸB to its corresponding DNA consensus sequence [6]. Helenalin, another inhibitor of active NF-ĸB, directly alkylates NF-ĸB p65, thereby inhibiting NF-ĸB binding to the respective DNA sequence [35]. Even we have no experimental evidence, the fact that both DMF and Helenalin contain electrophilic α, β-unsaturated carbonyl structures makes it likely that the modification of NF-κB which inhibits its DNA binding depends on alkylation. For Helenalin, it has been shown electrophilic α, β-unsaturated carbonyl structures react with nucleophilic protein sulfhydryl groups via a Michael-type addition [36, 37]. The NF-ĸB p65 and p50 contain a number of cysteine residues, therefore it is possible that DMF suppresses NF-ĸB/DNA binding by a direct interaction with sulfhydryl groups located in the DNA binding domain [38, 39].
Conclusion
In summary, our data show that DMF down-regulates PDGF-BB and TNF-α induced IL-6 secretion by human lung fibroblasts and their proliferation. Both effects may be mediated through the inhibition of NF-ĸB and AP-1 activity. The clinical efficacy of DMF and its long term safety profile in psoriasis makes DMF an interesting drug that could help to control inflammation and airway wall remodeling in inflammatory lung diseases.
Correspondence:
Michael Roth
University Hospital Basel, Department Research
Hebelstrasse 20
CH-4031 Basel
Switzerland
rothmic@uhbs.ch
References
1 Mrowietz, U, Asadullah, K. Dimethylfumarate for psoriasis: more than a dietary curiosity. Trends Mol Med. 2005;11:43–8.
2 Kappos L, Gold R, Miller DH, Macmanus DG, Havrdova E, Limmroth V, et al. Efficacy and safety of oral fumarate in patients with relapsing-remitting multiple sclerosis: a multicentre, randomised, double-blind, placebo-controlled phase IIb study. Lancet. 2008;372:1463–72.
3 Holgate ST. Pathogenesis of asthma. Clin Exp Allergy. 2008;38(6):872–97.
4 Sullivan DE, Ferris M, Nguyen H, Abboud E, Brody AR. TNF-alpha induces TGF-beta(1) expression in lung fibroblasts at the transcriptional level via AP-1 activation. J Cell Mol Med. 2009;13(8B):1866–76.
5 Tamm M, Bihl M, Eickelberg O, Stulz P, Perruchoud AP, Roth M. Hypoxia-induced interleukin-6 and interleukin-8 production is mediated by platelet-activating factor and platelet-derived growth factor in primary human lung cells. Am J Respir Cell Mol Biol. 1998;19(4):653–61.
6 Seidel P, Merfort I, Hughes JM, Oliver BG, Tamm M, Roth M. Dimethylfumarate inhibits NF-{kappa}B function at multiple levels to limit airway smooth muscle cell cytokine secretion. Am J Physiol Lung Cell Mol Physiol. 2009;297(2):L326–39.
7 McCormack PL, Lyseng-Williamson KA. Budesonide/Formoterol – A review of its use as maintenance and reliever inhalation therapy in asthma. Drugs. 2007;67:2407–31.
8 Adcock IM, Caramori G, Chung KF. New targets for drug development in asthma. Lancet. 2008;372(9643):1073–87.
9 Ammit AJ, Moir LM, Oliver BG, Hughes JM, Alkhouri H, Ge Q, et al. Effect of IL-6 trans-signaling on the pro-remodeling phenotype of airway smooth muscle. Am J Physiol. 2007;292:L199–L206.
10 Ray A, Tatter SB, May LT, Sehgal PB. Activation of the human “beta 2-interferon/hepatocyte-stimulating factor/interleukin 6” promoter by cytokines, viruses, and second messenger agonists. Proc Natl Acad Sci. USA. 1988;85(18):6701–5.
11 Roth M, Black JL. Transcription factors in asthma: are transcription factors a new target for asthma therapy? Curr Drug Targets. 2006;7(5):589–95.
12 Gesser B, Johansen C, Rasmussen MK, Funding AT, Otkjaer K, Kjellerup RB, et al. Dimethylfumarate specifically inhibits the mitogen and stress activated kinases 1 and 2 (MSK1/2): Possible role for its anti-psoriatic effect. J Invest Dermatol. 2007;127:2129–37.
13 Gerdes S, Shakery K, Mrowietz U. Dimethylfumarate inhibits nuclear binding of nuclear factor kappa B but not of nuclear factor of activated T cells and CCAAT/enhancer binding protein beta in activated human T cells. Br J Dermatol. 2007;156:838–42.
14 Vandermeeren M, Janssens S, Wouters H, Borghmans I, Borgers M, Beyaert R, et al. Dimethylfumarate is an inhibitor of cytokine-induced nuclear translocation of NFkappaB1, but not RelA in normal human dermal fibroblast cells. J Invest Dermatol. 2001;116:124–30.
15 Loewe R, Holnthoner W, Groge, M, Pillinger M, Gruber F, Hofer, E, et al. Dimethylfumarate inhibits TNF-induced nuclear entry of NF-kappa B/p65 in human endothelial cells. J Invest Dermatol. 2002;119:208.
16 Rehan VK, Asotra K, Torday JS. The effects of smoking on the developing lung: insights from a biologic model for lung development, homeostasis, and repair. Lung. 2009;187(5):281–9.
17 Plopper CG, Smiley-Jewell SM, Miller LA, Fanucchi MV, Evans MJ, Buckpitt AR, et al. Asthma/allergic airways disease: does postnatal exposure to environmental toxicants promote airway pathobiology? Toxicol Pathol. 2007;35(1):97–110.
18 Joad JP, Kott KS, Bric JM, Peake JL, Plopper CG, Schelegle ES, et al. Structural and functional localization of airway effects from episodic exposure of infant monkeys to allergen and/or ozone. Toxicol Appl Pharmacol. 2006;214(3):237–43.
19 Jenkins HA, Cool C, Szefler SJ, Covar R, Brugman S, Gelfand EW, et al. Histopathology of severe childhood asthma: a case series. Chest. 2003;124(1):32–41.
20 Racké K, Haag S, Bahulayan A, Warnken M. Naunyn Schmiedebergs Arch Pharmacol. 2008;378(2):193–201.
21 Nonaka M, Ogihara N, Fukumoto A, Sakanushi A, Kusama K, Pawankar R, et al. Combined Stimulation with Poly(I:C), TNF-alpha and Th2 Cytokines Induces TARC Production by Human Fibroblasts from the Nose, Bronchioles and Lungs. Int Arch Allergy Immunol. 2010;152(4):327–41.
22 Hardaker EL, Bacon AM, Carlson K, Roshak AK, Foley JJ, Schmidt DB, et al. Regulation of TNF-alpha- and IFN-gamma-induced CXCL10 expression: participation of the airway smooth muscle in the pulmonary inflammatory response in chronic obstructive pulmonary disease. FASEB J. 2004;18(1):191–3.
23 Eickelberg O, Pansky A, Koehler E, Bihl M, Tamm M, Hildebrand P, et al. Molecular mechanisms of TGF-(beta) antagonism by interferon (gamma) and cyclosporine A in lung fibroblasts. FASEB J. 2001;15(3):797–806.
24 Simon AR, Takahashi S, Severgnini M, Fanburg BL, Cochran BH. Role of the JAK-STAT pathway in PDGF-stimulated proliferation of human airway smooth muscle cells. Am J Physiol Lung Cell Mo Physiol. 2002;282:L1296-L1304.
25 Park D, Choi YB, Han MK, Kim UH, Shin J, Yun Y. Adaptor protein Lad relays PDGF signal to Grb2 in lung cells: a tissue-specific PDGF signal transduction. Biochem Biophys Res Commun. 2001;284(2):275–81.
26 Borger P, Miglino N, Baraket M, Black JL, Tamm M, Roth M. Impaired translation of CCAAT/enhancer binding protein alpha mRNA in bronchial smooth muscle cells of asthmatic patients. J Allergy Clin Immunol. 2009;123(3):639–45.
27 Roth M, Nauck M, Tamm M, Ziesche R, Block LH: Intracellular interleukin-6 modulates platelet-derived growth factor-induced proliferation of non-transformed cells. Proc Natl Acad Sci.USA,1995,92:1312–6.
28 Roth M, Nauck M, Yousefi S, Tamm M, Blaser K, Perruchoud AP, et al. Platelet-activating factor exerts mitogenic activity and stimulates expression of interleukin-6 and interleukin-8 in human lung fibroblasts via binding to its functional receptor. J Exp Med.1996;184:191–201.
29 Chiou YL, Shieh JJ, Lin, CY. Blocking of Akt/NF-kappa B signaling by pentoxifylline inhibits platelet-derived growth factor-stimulated proliferation in brown Norway rat airway smooth muscle cells. Pediatr Res. 2006;60:657–62.
30 Romashkova JA, Makarov SS. NF-kappa B is a target of AKT in anti-apoptotic PDGF signalling. Nature. 1999;401:86–90.
31 Rauch BH, Weber A, Braun M, Zimmermann N, Schrör K. PDGF-induced Akt phosphorylation does not activate NF-kappa B in human vascular smooth muscle cells and fibroblasts. FEBS Lett. 2000;481:3–7.
32 Barnes PJ. The cytokine network in asthma and chronic obstructive pulmonary disease. J Clin Invest. 2008;118(11):3546–56.
33 Sun SC, Ganchi PA, Ballard DW, Greene WC. NF-κB controls expression of inhibitor IκBα: evidence for an inducible autoregulatory pathway. Science. 1993;259:1912–5.
34 Kravchenko VV, Kaufmann GF, Mathison JC, Scott DA, Katz AZ, Grauer DC, et al. Modulation of gene expression via disruption of NF-kappaB signaling by a bacterial small molecule. Science. 2008;321(5886):259–63.
35 Lyss G, Knorre A, Schmidt TJ, Pahl HL, Merfort I. The anti-inflammatory sesquiterpene lactone helenalin inhibits the transcription factor NF-kappa B by directly targeting p65. J Biol Chem. 1998;273:33508–16.
36 Schmidt TJ, Ak M, Mrowietz U. Reactivity of dimethyl fumarate and methylhydrogen fumarate towards glutathione and N-acetyl-L-cysteine – preparation of S-substituted thiosuccinic acid esters. Bioorg Med Chem. 2007;15(1):333–42.
37 Schmidt TJ, Lyss G, Pahl HL, Merfort I. Helenanolide type sesquiterpene lactones. Part 5: the role of glutathione addition under physiological conditions. Bioorg Med Chem. 1999;7(12):2849–55.
38 Chen YQ, Ghosh S, Ghosh G. A novel DNA recognition mode by the NF-kappa B p65 homodimer. Nat Struct Biol. 1998;5:67–73.
39 Matthews JR, Wakasugi N, Virelizier JL, Yodoi J, Hay RT. Thioredoxin Regulates the Dna-Binding Activity of Nf-Chi-B by Reduction of A Disulfide Bond Involving Cysteine. Nucleic Acids Res. 1992;20:3821–30.