DOI: https://doi.org/10.4414/smw.2010.13112
With an age-adjusted incidence of 49 out of 100 000 per year, colorectal cancer is one of the most frequent malignant human tumours [1]. With regard to its pathogenesis, it is also one of the best understood. The evolution of colorectal cancer from polyps was first proposed by Morson [2]. Since the seminal publication of Vogelstein and Fearon [3], we understand that most colorectal cancers arise in a multistep process progressing from mucosal hyperplasia to adenomas and carcinomas. Many crucial molecular alterations underlying this tumour progression have been delineated. They include loss of common tumour suppressors such as APC (adenomatous polyposis coli), p53 and DPC4 (deleted in pancreatic cancer, Smad4), and activation of oncogenes such as Kras. However, until recently this knowledge has been irrelevant to cancer therapy. In 2008, the European Medicines Agency (EMEA) approved cetuximab, a monoclonal antibody directed against the epidermal growth factor receptor (EGFR), as a novel therapy for Kras wild-type (wt) metastatic colorectal cancer. At last the knowledge of colorectal tumour biology became useful for clinical decision-making. Since then our understanding of the signalling pathways involved in colorectal cancer progression has further increased, but in daily routine Kras remains the most important molecular marker for therapy-related decisions. The aim of this article is to review the molecular biology of Kras, to highlight the clinical consequences of Kras mutations and to discuss the role of Kras in metastatic colorectal cancer.
Human colorectal cancer develops through a series of genetic alterations. These genetic changes in intestinal epithelial cells lead first to mucosal hyperplasia and then to adenomas and carcinomas (the so-called adenoma-carcinoma sequence). These somatic mutations occur in a defined spatial and temporal manner, i.e., only mutations occurring in a specific cell type and at a defined time-point lead to the development of colorectal cancer. In a mouse model of intestinal cancer, APC deletion in the stem cell compartment of the colonic crypts causes the formation of adenomas, whereas the deletion of APC in short-lived cells outside the crypts does not [4]. In the model proposed by Vogelstein and Fearon [3], APC mutations are thought to occur very early in the development of colorectal cancer. Loss of APC results in decreased degradation of β-catenin and thereby activation of Wnt signalling and the expression of genes inducing cell proliferation and invasion (reviewed by [5]).
A second mutation that occurs rather early in adenomas is the activation of Kras (Kirsten rat sarcoma viral oncogene homologue). Kras is a small GTPase (guanosin triphosphate cleaving enzyme) involved in intracellular signal transduction. Importantly, it is the main transduction pathway for EGFR (epidermal growth factor receptor) signalling. A typical example of a mutation occurring late in the adenoma-carcinoma sequence is the inactivation ofp53, which relaxes cell cycle control and enables the tumour cells to evade apoptosis and continue proliferation. In addition, there is also evidence that mutant p53 can directly promote tumour cell invasion and metastasis [6].
The concept of the adenoma-carcinoma sequence has been verified in large patient series, and clinical evidence for the sequence is abundant. For example, the average age of patients with adenomas is 7–8 years lower than that of patients with colorectal cancer. Moreover, in surgical specimens benign adenomatous tissue can be found contiguous with cancerous lesions. In addition, 30% of patients with colorectal cancer also have synchronous adenomas, and metachronous adenomas develop in about the same proportion of patients after colorectal cancer has been diagnosed (reviewed by [7]). Nevertheless, it is probable that alternative pathways of carcinogenesis exist in colorectal cancer. One argument in favour of an alternative, non-polypoid origin of colorectal tumours is the fact that the anatomical distribution of adenomas and carcinomas is not identical. Histological analysis of colorectal cancers in patients and mouse models of human cancer has identified a group of flat tumours that most probably do not develop through a polypoid stage and may account for up to 10–20% of all colorectal cancers. Although they seem to be associated with more advanced pathologies, the frequency of mutations in Kras or in Braf, a serine/thereonine-protein kinase acting downstream of Kras, is the same as in the classical polypoid colorectal cancers [8]. The clinical relevance of non-polypoid carcinogenesis is a matter of ongoing debate and there is no final conclusion on this issue yet [9].
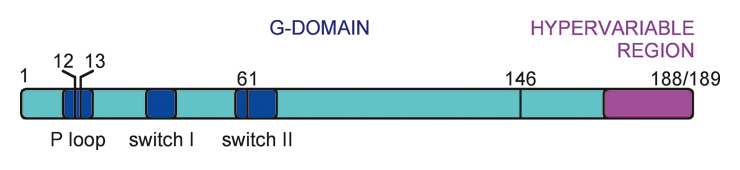
Kras is a small GTPase consisting of 188 or 189 amino acids (depending on the isoform). Somatic mutations in tumours occur at codons 12, 13, 61 or 146. Amino acids 12 and 13 are located in the GTP-binding P-loop, whereas amino acid 61 is situated in the switch-II region (fig. 1A). The switch regions control binding to Ras regulators and effectors. The G-domain of Kras is highly conserved. The hypervariable region controls membrane localisation through farnesylation and palmitoylation of amino acid residues. Kras is found mutated in colorectal (30–60%), pancreatic (60%), biliary (33%), ovarian (17%) and endometrial cancer (15%). In lung cancer Kras mutations occur in 30–40% of adenocarcinomas but are infrequent in other histologies and in non-smokers [10]. In rare cases Kras mutations are found in bladder, breast, cervical, kidney, liver and thyroid cancer as well as in melanomas and myeloid leukaemia [11]. Ras mutations found in human tumours usually result in a severe reduction of GTPase activity and are thus constitutive in activating Ras signalling [12] (fig. 1B). In experimental systems, mutant Kras is capable of transforming adult somatic cells of a variety of organs and thus of inducing tumour formation [13].
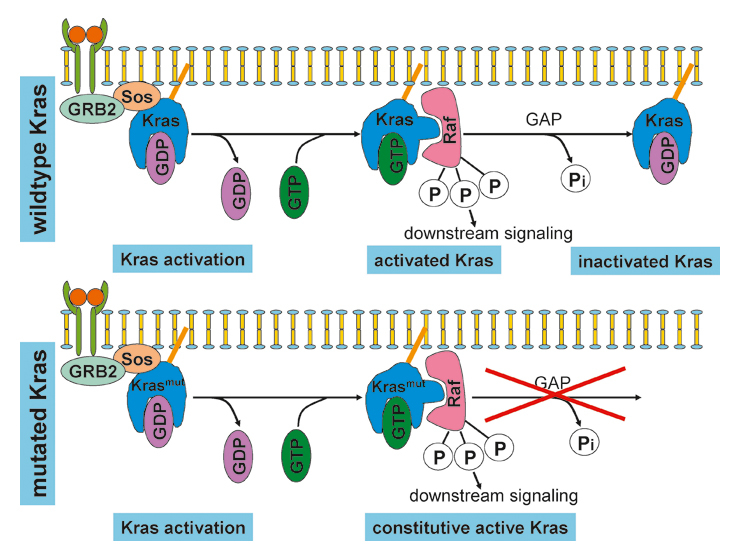
Figure 1
Kras: structure and activation. (A) The Kras gene. Mutations in human tumours occur most commonly in codons 12 or 13 but mutations in codons 61 and 146 have been described. (B) Kras activation. Following the activation of a receptor tyrosine kinase (RTK), a complex containing the activated receptor, GRB2 (growth factor receptor-bound protein 2) and Sos (son of sevenless) is formed on the cytosolic face of the cell membrane. Binding of Sos to Kras-GDP leads to a conformational change of the switch 1 and 2 regions, thereby mediating the exchange of GDP for GTP. This triggers a second conformational change that allows Ras-GTP to bind to and activate Raf. In wild-type Kras (top panel), signalling is terminated by hydrolysis of GTP to GDP. In mutated Kras (bottom panel), the GTPase activity of Kras is severely impaired, making hydrolysis of GTP and termination of signalling impossible. GEF = GTP exchange factor. GAP = GTPase activating proteins.
In colorectal cancer, mutations of Kras occur almost exclusively in exon 2, usually involving codons 12 or 13. Most studies conducted in patients with colorectal cancer have therefore focused on sequencing these two codons [14]. However, mutations in codons 61 and 146 have been described and may account for up to 10% of patients bearing Kras-mutant tumours [15]. The two most frequent Kras mutations in microsatellite-stable tumours are G12D and G12V. In microsatellite-instable colorectal cancer and in hereditary non-polyposis coli cancer (HNPCC), G12D and G13D mutations are most common [16]. How these different Kras mutations may specifically modulate intracellular signalling is not well understood and remains a matter of ongoing research.
The mutations discussed above occur in coding regions of the Kras gene. Recent experimental evidence has uncovered the presence of mutations in the 3’-untranslated region (3’-UTR) of Kras. 3’-UTR mutations have been associated with poor prognosis in colorectal cancer patients with cetuximab/irinotecan salvage therapy [17]. Interestingly, poly-ADP-ribose polymerase 1 (PARP1) binds directly to the murine Kras gene promoter and thus upregulates Kras expression and Kras signalling [18].
In patients with adenocarcinomas of the colon or the rectum Kras is mutated in about 30–60% of the cases. The frequency of Kras mutations does not depend on the TNM or Dukes stage of the carcinoma and the same mutation rate is found in familial colorectal cancer. Compared with carcinomas, approximately the same fraction of adenomas larger than 1 cm also harbour Kras mutations, whereas the rate drops to 10–15% in adenomas smaller than 1 cm. This underlines the notion that Kras is involved in carcinogenesis and that Kras mutation is an early event in colorectal cancer [19].
Many studies have investigated the concordance between Kras mutations in the primary tumour and in related metastases. Concordance has been found to range between 68 and 100%, and recent data indicate that it probably lies between 92 and 96% in an unselected population of patients with metastatic colorectal cancer [20–27]. However, metachronous single or multiple mutations of Kras in metastases from Kras-wild-type primary tumours have been reported and may, in some instances, account for acquired resistance against anti-EGFR-antibody therapy [28].
Kras is a central player in intracellular signalling (fig. 2). It may be activated by the EGF receptor or possibly other receptor tyrosine kinases. Through interaction with phosphatidylinositol-3 kinase (PI3K) and activation of the downstream effectors such as mammalian target of rapamycin (mTOR), Kras indirectly modulates cell survival. Through the Braf/mitogen-activated kinase (MAPK) pathway it also influences cell proliferation (reviewed by [29]).
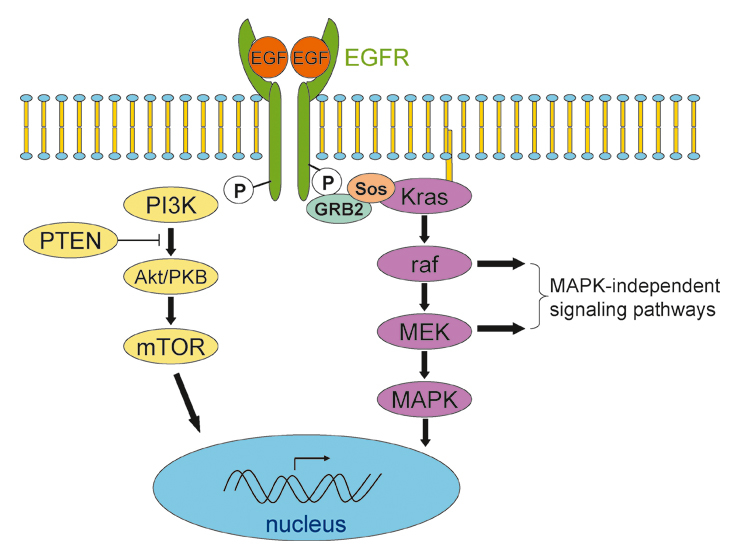
Figure 2
Principal signalling pathways activated by the EGF receptor. Activated EGF receptors dimerise and start signalling through the PI3K-AKT/PKB-mTOR and the Kras-Braf-MEK pathway. Activation of Kras and Braf triggers not only MAPK signalling but also MAPK-independent signal transduction.
Whereas the relevance of Kras mutations in the pathogenesis of colorectal cancer is undisputed, the data regarding the role of Kras in tumour progression are conflicting. On the one hand, the frequency of Kras mutations has been reported to be independent of the tumour stage [19]. On the other hand, in a Spanish cohort of 230 patients the mutation rate of Kras in the primary tumour was 35% if no lung metastases were present, while the probability of Kras mutations increased to 57% in patients with lung metastases [20]. In an American study including patients with colorectal cancer and liver metastases, the presence of Kras mutations in the metastases was an independent predictor of poor survival after resection of liver metastases [30].
Preclinical data indicate that there are different mechanisms through which Kras may promote tumour progression and metastasis formation. Not surprisingly, oncogenic Ras can promote tumour cell proliferation through Braf and mitogen-activated protein kinase kinase (MEK) signalling (fig. 2) [31]. Nevertheless, the extent to which neoplastic proliferation occurs is dependent on the cellular context [32]. Interestingly, in colorectal cancer cell lines and mouse intestinal epithelial cells, Kras, Braf and MEK signalling do not necessarily lead to MAPK activation. It therefore seems probable that oncogenic Kras, Braf and MEK signalling occurs, at least in part, through MAPK-independent pathways [33] (fig. 2).
Although the preclinical data are quite robust, it is less appreciated that mutant Ras is also capable of stimulating angiogenesis through induction of interleukin-8 (IL-8) synthesis [34]. Expression of mutant Kras in murine fibroblasts is sufficient to up-regulate genes involved in proliferation and angiogenesis [35]. Finally, activated Kras inhibits DNA repair-related genes in colonic crypts in a 3-dimensional culture system [36]. One can easily imagine that the combination of tumour cell proliferation, induction of angiogenesis and inhibition of DNA repair is a potent mechanism for enhancement of tumour progression and metastasis. However, in contrast to these preclinical results, available clinical data are somewhat less convincing and further studies are warranted.
The guidelines of the American Society of Clinical Oncology (ASCO) state that all patients with metastatic colorectal cancer who are candidates for anti-EGFR-antibody therapy should be tested for Kras mutations in the tumours. If a mutation is detected in codons 12 or 13, these patients should not receive anti-EGFR antibodies as part of their treatment [14]. Analysis of the primary tumour for Kras mutations seems sufficient, since the concordance between Kras mutations in the primary tumour and mutations in related metastases is very high [20]. Kras mutations can be detected in formalin-fixed surgical or biopsy tumour samples embedded in paraffin [37]. If paraffin-embedded samples are not available, Kras testing may be performed on cytological specimen. In a small study with 19 patients, concordance between formalin-fixed paraffin-embedded samples and cytological samples was 18/19 [38]. In another study of 76 patients, Kras mutations were detected in circulating tumour cells isolated from peripheral blood in 30 out of 33 patients with Kras mutations in the primary tumour [39]. Detection of mutant DNA in the peripheral blood can be used not only to search for Kras mutations, it is also a useful tool to monitor tumour dynamics in patients with colorectal cancer [40]. Mutations in the DNA of colorectal cancers, including mutant Kras, can also be detected in stool. In a clinical study, 23 of the 25 stool DNA samples analysed contained mutations that were present in the corresponding tumours from the same patients [41]. However, from a clinical perspective determination of Kras mutations in the primary tumour remains the most reliable technique and should therefore be considered the standard procedure for the time being.
Several targeted therapies against EGFR signalling activities have been established. Cetuximab is a human/mouse chimaeric IgG1 monoclonal antibody that binds to the extracellular domain of the EGFR and inhibits EGFR-mediated signalling [42]. Panitumumab is a fully human IgG2 monoclonal antibody targeting EGFR (reviewed by [43]). Initial evaluation of cetuximab as monotherapy in patients with EGFR-expressing chemotherapy-refractory tumours yielded response rates of approximately 10%. Detection of EGFR expression by immunohistochemistry was not sufficient to predict response to EGFR blocking antibodies. Hence, based on the knowledge of EGFR signalling, downstream effectors of the EGFR pathway have been investigated as potential surrogate markers of cetuximab therapy response. Activating mutations of Kras induce constitutive Kras-Braf-MEK signalling, which cannot be suppressed by inhibition of EGFR. Thus, one of the first surrogate markers to be investigated was Kras [44]. In a well-designed study, Karapetis et al. have shown that patients with activating Kras mutations did not benefit from anti-EGFR-antibody therapy [45]. In this trial, 572 patients with metastatic colorectal cancer were randomly assigned to either cetuximab plus best supportive care or best supportive care alone. Cetuximab was given in a dose of 400 mg/m2 at day one and then weekly in a dose of 250 mg/m2. Cetuximab was continued until disease progression or until the patient could not tolerate the toxic effects. 394 patients were retrospectively evaluated for Kras mutational status. In patients with wild-type Kras the median survival on cetuximab monotherapy in comparison to best supportive care was increased from 4.8 to 9.5 months, whereas there was no difference in overall survival in patients with activating Kras mutations [45]. Based on this study, anti-EGFR-antibody therapy has been approved in patients suffering from metastatic colorectal cancer with unmutated Kras. Combining the cytotoxic agents irinotecan or oxaliplatin with anti-EGFR-antibodies in metastatic colorectal cancer is only effective in Kras-wild-type tumours, a notion that has been confirmed by a further analysis of the data obtained from the CRYSTAL and OPUS trials [46–48]. Importantly, recent data have raised concern about a possible detrimental effect of anti-EGFR-antibody therapy in Kras-mutated colorectal cancer. In the PRIME trial, patients received either Folfox or Folfox plus panitumumab as first line therapy for metastatic cancer. Patients with wild-type Kras had better progression-free survival (PFS) if treated with panitumumab, yet PFS was inferior if patients with mutated Kras received anti-EGFR-antibody therapy in addition to Folfox [49]. This underlines the necessity of timely Kras testing in colorectal cancer. Taken together, there is sound clinical evidence in favour of anti-EGFR-antibody therapy in wild-type Kras colorectal cancer. However, in the light of new data from recent studies such as the British COIN trial, the efficacy of anti-EGFR-antibody therapy may vary according to the type, schedule and administration route of the accompanying chemotherapy [50].
Although Kras mutations are clearly predictive for response to anti-EGFR-antibody therapy, there are conflicting results concerning their prognostic relevance in early and advanced colorectal cancer. Recent data indicate that Kras mutations do not influence prognosis [51]. In addition, Kras mutations do not predict response to treatment with oxaliplatin or irinotecan [52].
In summary, anti-EGFR-therapy improves OS in patients with metastatic colorectal cancer and wild-type Kras. Testing is necessary, since anti-EGFR-therapy in patients with mutated Kras may not only be ineffective but also detrimental.
Kras mutations account for 30–40% of colorectal cancer patients who are not responsive to anti-EGFR-antibody therapy. This finding has motivated a search for other components of the MAPK or the PI3K pathway that might provide additional predictive information. Braf is a cytoplasmatic kinase which is activated immediately downstream of Kras and therefore seems to represent a suitable target when searching for other predictive markers (fig. 2). Notably, mutant Braf V600E induces constitutive Braf-MEK signalling, similar to the effect of activating Kras mutations, and Kras and Braf mutations seem mutually exclusive [53]. This may be due to reduced survival and proliferation of cells concomitantly bearing Kras and Braf mutations. However, combined mutations do occasionally occur in tumour cells capable of escaping apoptosis, and these cells may be observed in association with an aggressive, pro-angiogenic tumour phenotype [54]. Braf mutations occur more often in mismatch repair-deficient cancers, but mutations in premalignant lesions and mismatch repair-proficient colorectal tumours have been described as well [53]. The concordance between Braf status in the primary tumour and in related metastases is high in wild-type Braf tumours (>90%), whereas it is low in primary colorectal cancers with mutated Braf [55].
Braf may be both prognostic and predictive for response to treatment with anti-EGFR-antibodies [56]. However, a recent retrospective analysis of pooled data from the OPUS and CRYSTAL trials does not support the notion that Braf mutations have a predictive value [57]. However, the sample size in the latter analysis was very small. Thus, in the absence of reliable prospective data, the question of Braf mutations and their predictive significance is unresolved and further studies will be necessary to clarify the issue.
Whether EGFR gene amplification is a valuable predictor for a response to anti-EGFR therapy is still fiercely debated. The conflicting data are probably due to both technical difficulties in determining the exact copy number and to confounding factors such as downstream mutations of the MAPK signalling pathway. A recent study has considered these factors and has found a weak but statistically significant correlation between EGFR gene copy number and response to anti-EGFR-antibody therapy [58].
Mutational analysis of the tumour suppressor gene PTEN in patients with colorectal cancer has shown that mutations of PTEN are prognostically relevant. However, there is still debate as to whether they predict response to anti-EGFR-antibody therapy [58–60]. Concerning the role of PI3K, the data are conflicting as to whether mutations of the catalytic subunit of class 1A PI3K (PIK3CA) have an impact on tumour response to anti-EGFR-antibody therapy [59, 61]. These contradictory results may at least in part be due to the fact that we still do not possess a reliable and readily available test to indicate activation of the EGFR pathway. In the future this problem may be tackled, for instance, using serum proteomics, thereby identifying clinically significant tumour-dependence on EGFR signalling [62].
When it became clear that Kras mutations precluded a beneficial effect of anti-EGFR-antibodies, attention shifted to blocking EGFR signalling downstream of Kras, in particular on the level of Braf. Indeed, first-generation Braf inhibitors can suppress proliferation of Braf mutant cell lines in vitro and in vivo. However, they are unexpectedly ineffective against Kras mutant cells and cannot block MAPK/Erk activation in these cells [63]. Paradoxically, blocking one ATP-binding site of dimeric Braf stimulates the kinase activity of the other. This occurs only in cells bearing mutant Kras and wild-type Braf. However, this is a setting found in 30–60% of colorectal cancer patients and thus represents a serious obstacle to successful therapy. Different mechanisms have been proposed to explain the phenomenon but further research is required before the EGFR-induced signalling cascade downstream of Kras can be successfully blocked in the clinic [63–65].
The EGFR signalling pathway is a major player in colorectal tumourigenesis and tumour progression. Kras mutations mimic continuous EGFR signalling activation and thus induce tumour cell proliferation and tumour angiogenesis.
All patients with colorectal cancer who may qualify for anti-EGFR-antibody treatment should be tested for Kras mutations. Patients with Kras-wild-type tumours benefit from therapy with anti-EGFR-antibodies, while patients with mutant Kras do not. In patients with metastasised colorectal cancer and wild-type Kras, a first line therapy with Folfox or Folfiri in combination with cetuximab or panitumumab is a good option. The addition of anti-EGFR-antibody therapy to standard Folfox or Folfiri regimens increases response rates, progression-free survival and overall survival (OS) in this population. In patients with mutated Kras, anti-EGFR-antibody therapy should be avoided. As an alternative, therapy with Folfox or Folfiri in combination with bevacizumab may be considered.
In the future, we will need better predictive biomarkers in order to choose the best therapy for the patients, and we will depend on new therapeutic targets both within and outside the setting of EGFR and Kras signalling. However, understanding Kras is a first and important step towards individualised therapy of colorectal cancer.
We apologise to all colleagues whose important work we could not cite due to space restrictions.
1 U.S. National Cancer Institute. SEER database, 2002–2006.
2 Morson B. President’s address: The polyp-cancer sequence in the large bowel. Proc R Soc Med. 1974;67:451–7.
3 Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, et al. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319(9):525–32.
4 Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009;457:608–12.
5 Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer. 2008;8(5):378–98.
6 Muller PA, Caswell PT, Doyle B, Iwanicki MP, Tan EH, Karim S, et al. Mutant p53 drives invasion by promoting integrin recycling. Cell. 2009;139(7):1327–41.
7 Leslie A, Carey FA, Pratt NR, Steele RJC. The colorectal adenoma-carcinoma sequence. Br J Surg. 2002;89(7)845–60.
8 Uronis JM, Herfarth HH, Rubinas TC, Bissahoyo AC, Hanlon K, Threadgill DW. Flat colorectal cancers are genetically determined and progress to invasion without going through a polypoid stage. Cancer Res. 2007;67(24):11594–600.
9 Smith D, Ballal M, Hodder R, Selvachandran SN, Cade D. The adenoma carcinoma sequence: an indoctrinated model for tumorigenesis, but is it always clinical reality? Colorectal disease. 2006;8:296–301.
10 Berns A. Cancer. Improved mouse models. Nature. 2001;410(6832):1043–4.
11 Bamford S, Dawson E, Forbes S, Clements J, Pettett R, Dogan A, et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br J Cancer. 2004;91(2):355–8.
12 Gibbs JB, Sigal IS, Poe M, Scolnick EM. Intrinsic GTPase activity distinguishes normal and oncogenic ras p21 molecules. Proc Natl Acad Sci USA. 1984;81(18):5704–8.
13 Gidekel Friedlander SY, Chu GC, Snyder EL, Girnius N, Dibelius G, et al. Context-dependent transformation of adult pancreatic cells by oncogenic K-Ras. Cancer Cell. 2009;16:379–89.
14 Allegra CJ, Jessup M, Somerfield MR, Hamilton SR, Hammond EH, Hayes DF, et al. American society of clinical oncology provisional clinical opinion: testing for Kras gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J Clin Oncol. 2009;27:2091–6.
15 Loupakis F, Ruzzo A, Cremolini C, Vincenzi B, Salvatore L, Santini D, et al. Kras codon 61, 146 and Braf mutations predict resistance to cetuximab plus irinotecan in Kras codon 12 and 13 wild-type metastatic colorectal cancer. Br J Cancer. 2009;101(4):715–21.
16 Oliveira C, Westra JL, Arango D, Ollikainen M, Domingo E, Ferreira A, et al. Distinct patterns of Kras mutations in colorectal carcinomas according to germline mismatch repair defects and hMLH1 methylation status. Hum Mol Genetics. 2004;13(19):2303–11.
17 Graziano F, Canestrari E, Loupakis F, Ruzzo A, Galluccio N, Santini D, et al. Pharmacogenomics J. 2010;AOP:doi:10.1038/tpj.2010.9
18 Cogoi S, Paramasivam M, Membrino A, Yokoyama K, Xodo L. The Kras promoter responds to myc-associated zinc finger and poly-ADP-ribose polymerase 1 proteins which recognize a critical quadruplex-forming GA-element. J Biol Chem. 2010:AOP:doi/10.1074/jbc.M110.101923
19 Brink M, de Goeij AFPM, Weijenberg MP, Roemen GMJM, Lentjes MHFM, Pachen MMM. K-ras oncogene mutations in sporadic colorectal cancer in The Netherlands Cohort Study. Carcinogenesis. 2003;24(4):703–10.
20 Cejas P, López-Gómez M, Aguayo C, Madero R, de Castro Carpeño J, Belda-Iniesta C, et al. KRAS Mutations in Primary Colorectal Cancer Tumors and Related Metastases: A Potential Role in Prediction of Lung Metastasis. PLoS ONE. 2009;4(12):e8199.doi:10.1371/journal.pone.0008199
21 Molinari F, Martin V, Saletti P, De Dosso S, Spitale A, Camponovo A, et al. Differing deregulation of EGFR and downstream proteins in primary colorectal cancer and related metastatic sites may bei clinically relevant. Br J Cancer. 2009;100:1087–94.
22 Artale S, Sartore-Bianchi A, Veronese SM, Gambi V, Sartnataro CS, Siena S. Mutations of Kras and Braf in primary and matched metastatic sites of colorectal cancer. J Clin Oncol. 2008;26(25):4217–9.
23 Santini D, Loupakis F, Vincenzi B, Foriani I, Stasi I, Canestrari E, et al. High concordance of Kras status between primary colorectal tumors and related metastatic sites: implications for clinical practice. Oncologist. 2008;13(12):1270–5.
24 Zauber P, Sabbath-Solitare M, Marotta SP, Bishop DT. Molecular changes in the Ki-ras and APC genes in primary colorectal carcinoma and synchronous metastases compared with the findings in accompanying adenomas. Mol Pathol. 2003;56(3):137–40.
25 Etienne-Grimaldi MC, Formento JL, Francoual M, Francois E, Formento P, Renée N, et al. Kras mutations and treatment outcome in colorectal cancer patients receiving exclusive fluoropyrimidine therapy. Clin Cancer Res. 2008;14(15):4830–5.
26 Suchy B, Zietz C, Rabes HM. Kras point mutations in human colorectal carcinomas: relation to aneuploidy and metastasis. Int J Cancer. 1992;52(1):30–3.
27 Oliveira C, Velho S, Moutinho C, Ferreira A, Preto A, Domingo E, et al. Kras and Braf oncogenic mutations in MSS colorectal cancer progression. Oncogene. 2007;26(1):158–63.
28 Bouchahada M, Karaoué A, Saffroy R, Innominato P, Gorden L, Guettier C, et al. Acquired Kras mutations during progression of colorectal cancer metastases: possible implications for therapy and prognosis. Cancer Chemother Pharmacol. 2010;DOI10.1007/s00280-010-1298-9
29 Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer. 2007;7:295–308.
30 Nash GM, Gimbel M, Shia J, Nathanson DR, Ndubuisi MI, Zeng ZS, et al. Kras mutation correlates with accelerated metastatic progression in patients with colorectal liver metastasis. Ann Surg Oncol. 2009;doi:10.1245/s10434-009-0605-3
31 Tuveson DA, Shaw AT, Willis NA, Silver DP, Jackson EL, Chang S, et al. Endogenous oncogenic Kras (G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell. 2004;5(4):375–87.
32 Guerra C, Mijimolle N, Dhawahir A, Dubus P, Barradas M, Serrano M, et al. Tumor induction by an endogenous Kras oncogene is highly dependent on cellular context. Cancer Cell. 2003;4(2):111–20.
33 Haigis KM, Kendall KR, Wang Y, Cheung A, Haigis MC, Glickman JN, et al. Nat Genetics. 2008;40(5):600–8.
34 Sparmann A, Bar-Sagi D. Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell. 2004;6(5):447–58.
35 Horsch M, Recktenwald CV, Schädler S, Hrabé de Angelis M, Seliger B, Beckers J. Overexpressed vs mutated Kras in murine fibroblasts: a molecular phenotyping study. Br J Cancer. 2009;100(4):656–62.
36 Tsunoda T, Takashima Y, Fujimoto T, Koyanagi M, Yoshida Y, Doi K, et al. Three-dimensionally specific inhibition of DNA repair-related genes by activated Kras in colon crypt model. Neoplasia. 2010;12(5):397–404.
37 Weichert W, Schewe C, Lehmann A, Sers C, Denkert C, Budczies J, et al. Kras genotyping of paraffin-embedded colorectal cancer tissue in routine diagnostics. J Mol Diagn. 2010;12(1):35–42.
38 Troncone G, Malapelle U, Cozzolino I, Palombini L. Kras mutation analysis on cytological specimens of metastatic colorectal cancer. Diag Cytopathol. 2010;0:doi:10.1002/dc
39 Yen LC, Yeh YS, Chen CW, Wang HM, Tsai HL, Lu CY, et al. Detection of Kras oncogene in peripheral blood as predictor of the response to cetuximab plus chemotherapy in patients with metastatic colorectal cancer. Clin Cancer Res. 2009;15(13):4508–13.
40 Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008;14(9):985–90.
41 Diehl F, Schmidt K, Durkee KH, Moore KJ, Goodman SN, Shuber AP, et al. Analysis of mutations in DNA isolated from plasma and stool of colorectal cancer patients. Gastroenterology. 2008;135:489–98.
42 Prewett M, Rockwell P, Rockwell RF, Giorgio NA, Mendelsohn J, Scher HI, et al. The biologic effects of C225, a chimeric monoclonal antibody to the EGFR, on human prostate carcinoma. J Immunother Emphasis Tumor Immunol. 1996;19(6):419–27.
43 Peeters M, Balfour J, Arnold D. Panitumumab – a fully human anti-EGFR monoclonal antibody for treatment of metastatic colorectal cancer. Aliment Pharmacol Therapeutics. 2008;28:269–81.
44 Lièvre A, Bachet JB, Le Corre D, Boige V, Landi B, Emile JF, et al. Kras mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66(8):3992–5.
45 Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Dongsheng T, Tebbutt NC, et al. Kras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359(17):1757–65.
46 Van Cutsem E, Rougier P, Köhne C. A meta-analysis of the CRYSTAL and OPUS studies combining cetuximab with chemotherapy (CT) as 1st-line treatment for patients (pts) with metastatic colorectal cancer (mCRC): Results according to Kras and Braf mutation status. Eur J Cancer. 2009;7(S345):abstr6077.
47 Van Cutsem E, Köhne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009;360(14):1408–17.
48 Tabernero J, Van Cutsem E, Diaz-Rubio E, Cervantes A, Humblet Y, André T, et al. Phase II trial of cetuximab in combination with fluorouracil, leucovorin, and oxaliplatin in the first-line treatment of metastatic colorectal cancer. J Clin Oncol. 2007;25(33):5225–32.
49 Siena S, Cassidy J, Tabernero J, Burkes ME, Barugel Y, Humblet D, et al. Randomized phase III study of panitumumab (pmab) with FOLFOX4 compared to FOLFOX4 alone as first-line treatment (tx) for metastatic colorectal cancer (mCRC): PRIME trial. ASCO GI Cancers Symposium 2010; Abstract 283.
50 Maughan TS, Adams R, Smith CG, Seymour T, Wilson RH, Meade AM, et al. Oxaliplatin and fluoropyrimidine chemotherapy plus or minus cetuximab: The effect of infusional 5-FU or capecitabine on the outcomes of the MRC COIN trial in advanced colorectal cancer. ASCO GI Cancers Symposium, 2010; Abstract 402.
51 Roth AD, Tejpar S, Delorenzi M, Yan P, Fiocca R, Klingbiel D, et al. Prognostic role of Kras and Braf in Stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 Trial. J Clin Oncol. 2010;28(3):466–74.
52 Richman SD, Seymour MT, Chambers P, Elliott F, Daly CL, Meade AM, et al. Kras and Braf mutations in advanced colorectal cancer are associated with poor prognosis but do not preclude benefit from oxaliplatin or irinotecan: results from the MRC FOCUS trial. J Clin Oncol. 2009;27(35):5931–7.
53 Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. RAF/RAS oncogenes and mismatch-repair status. Nature. 2002;418(29):934.
54 Monticone M, Biollo E, Maffei M, Donadini A, Romeo F, Storlazzi CT, et al. Gene expression deregulation by Kras G12D and G12V in a Braf V600E context. Molecular Cancer. 2008;7(92):doi:10.1186/1476-4598-7-92
55 Santini D, Spoto C, Loupakis F, Vincenzi B, Silvestris N, Cremolini C, et al. High concordance of BRAF status between primary colorectal tumours and related metastatic sites: implications for clinical practice. Ann Oncol. 2010;21(7):1565.
56 Di Nicolantonio F, Martini M, Molinari F, Sartore-Bianchi A, Arena S, Saletti P, et al. Wild-type Braf is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol. 2008;26(35):5705–12.
57 Bokemeyer C, Kohne C, Rougier P, Stroh C, Schlichting M, Van Cutsem V. Cetuximab with chemotherapy (CT) as 1st-line treatment for metastatic colorectal cancer (mCRC): Analysis of the CRYSTAL and OPUS studies according to Kras and Braf mutation status. J Clin Oncol. 2010;28:7s(suppl; abstr 3506).
58 Laurent-Puig P, Cayre A, Manceau G, Buc E, Bachet JP, Lecomte T, et al. Analysis of PTEN, Braf, and EGFR status in determining benefit from cetuximab therapy in wild-type Kras metastatic colon cancer. J Clin Oncol. 2009;27(35):5924–30.
59 Sartore-Bianchi A, Martini M, Molinari F, Veronese S, Nichelatti M, Artale S, et al. PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res. 2009;69(5):1851–7.
60 Loupakis F, Pollina L, Stasi I, Ruzzo A, Scartozzi M, Santini D, et al. PTEN expression and Kras mutations on primary tumors and metastases in the prediction of benefit from cetuximab plus irinotecan for patients with metastatic colorectal cancer. J Clin Oncol. 2009;27(16):2622–9.
61 Prenen H, De Schutter J, Jacobs B, De Roock W, Biesmans B, Claes B, et al. PIK3CA mutations are not a major determinant of resistance to the epidermal growth factor receptor inhibitor cetuximab in metastatic colorectal cancer. Clin Cancer Res. 2009;15(9):3184–8.
62 Chung CH, Seeley EH, Roder H, Grigorieva J, Tsypin M, Roder J, et al. Detection of tumor epidermal growth factor receptor pathway dependence by serum mass spectrometry in cancer patients. Cancer Epidemiol Biomarkers Prev. 2010;19(2):OF1-8, doi: 10.1158/1055-9965.EPI-09-0937
63 Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, et al. Kinase-dead Braf and oncogenic Ras cooperate to drive tumor progression through Cras. Cell. 2010;140(2):209–21.
64 Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, et al. Nature. 2010;AOPdoi10.1038/nature08833
65 Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. Nature. 2010;AOPdoi10.1038/nature08902
Research in the laboratory of the authors related to this review article has been supported by the EU-FP6 framework programme BRECOSM LSHC-CT-2004-503224, the EU-FP7 framework programme TuMIC 2008-201662, the NCCR Molecular Oncology of the Swiss National Science Foundation, the Swiss Cancer League, the Krebsliga beider Basel, the Gebert-Rüf Foundation and the Desirée and Nils Yde Foundation.