Inactivation of the hypermethylated in cancer 1 tumour suppressor – not just a question of promoter hypermethylation?
DOI: https://doi.org/10.4414/smw.2010.13106
M
Jenal, C
Britschgi, MF
Fey, MP
Tschan
Summary
The chromosomal region 17p13.3 is frequently deleted or epigenetically silenced in a variety of human cancers. It includes the hypermethylated in cancer 1 (HIC1) gene placed telomerically to the p53 tumour suppressor gene. HIC1 encodes a transcriptional repressor, and its targets identified to date are genes involved in proliferation, tumour growth and angiogenesis. In addition, HIC1 functionally cooperates with p53 to suppress cancer development.
Frequent allelic loss at position 17p13.1 in human cancers often points to mutations of the tumour suppressor p53. However, in a variety of cancer types, allelic loss of the short arm of chromosome 17 may hit regions distal to p53 and, interestingly, without leading to p53 mutations. Furthermore, the neighbouring region 17p13.3 often shows loss of heterozygosity or DNA hypermethylation in various types of solid tumours and leukaemias. In line with this concept, Wales et al. described a new potential tumour suppressor in this region and named it hypermethylated in cancer 1 (HIC1). Further, it was shown that in the majority of cases hypermethylation of this chromosomal region leads to epigenetic inactivation of HIC1.
A role for HIC1 in tumour development is further supported by a mouse model, since various spontaneous, age- and gender-specific malignant tumours occur in heterozygous Hic1
+/– knockout mice. Furthermore, exogenously delivered HIC1leads to a significant decrease in clonogenic survival in cancer cell lines. This review highlights the role of HIC1 inactivation in solid tumours and particularly in leukaemia development.
HIC1 gene structure and regulation
There are conflicting reports in the literature about the HIC1 genomic nomenclature. Our review will confine itself to the most frequently used nomenclature proposed by Carter et al. [1]. Up to now six HIC1 transcripts (1a-f) have been described which are generated from different promoters through alternative splicing events (fig. 1A). The HIC1 transcripts 1a, 1b and 1c were detected in various normal tissues with strong predominance of the exon 1a transcript [2, 3]. For the HIC1 transcripts 1d and 1e, which yield from an additional alternative splice event in exon 1c, no expression data are available. Recently, Mondal et al. discovered a further unspliced HIC1 transcript 1f in human leukocytes. The authors stated that transcript 1f may play a role in modulating HIC1 protein levels in cancer cells [4].
The HIC1 protein is a sequence-specific 714 amino acid transcriptional repressor with five Krüppel-like zinc fingers in its C-terminal part, and an N-terminal broad complex, tramtrack, and bric à brac/poxviruses and zinc-finger (BTB/POZ) protein-protein interaction domain (fig. 1B) [5–7]. The zinc fingers are needed for DNA binding, whereas the BTB/POZ domain is important for dimerisation and transcriptional repression [5]. A second repression domain was found in the central region which may recruit the C-terminal binding proteins (CtBPs) for repression [8, 9]. Deltour et al. [8] demonstrated that HIC1 mediates transcriptional repression by both HDAC-dependent and -independent mechanisms.
At present there are two known transcriptional regulators of HIC1. A potential role has been suggested for the tumour suppressor p53 in activating transcription of HIC1 [2, 7]. Later we identified a p53 responsive element (PRE) 500bp upstream of the TATA-box containing promoter P0 [10]. We demonstrated that this PRE is necessary and sufficient to mediate induction of transcription by p53. Moreover, knockdown of p53 prevented HIC1mRNA induction in response to UV-induced DNA damage. Interestingly, other members of the p53 family, for example TAp73α/β and ΔNp63α, also induced HIC1 transcription via PRE. We recently identified E2F1 as a novel, positive regulator of HIC1[11]. E2F1 is a transcripton factor involved in different cellular processes such as cell cycle progression, DNA replication, oncogenic transformation and apoptosis responses. E2F1 may promote cell growth or cell death depending on the amount of active E2F1 in the cell and the cellular context [12]. We showed that E2F1 binds the HIC1 promoter in vivo, and identified two E2F-responsive elements in the proximal HIC1 promoter which are necessary to mediate induction of HIC1 transcription by E2F1. When we induced DNA damage by treating cells with etoposide, HI
C
1 was upregulated. This effect was dependent on E2F1, since E2F1knockdown by RNA interference diminished HIC1induction. We therefore propose a role for the E2F1-HIC1 pathway in the DNA damage response.
On the other hand, several posttranslational regulatory mechanisms have been described for HIC1: (a) glycosylation of the HIC1 protein preferentially occurs in the DNA-binding domain but does not affect its specific DNA-binding activity; (b) sumoylation of the conserved lysine K314 in the central region was found to reduce HIC1 repressive activity; and (c) acetylation of the same K314 residue affected HIC1 transcriptional activity (reviewed in [13]).
Possible HIC1 tumour suppressor pathways
The list of known HIC1 targets is steadily growing, and currently there are eight confirmed, transcriptionally repressed HIC1 targets as well as one target that is inactivated by HIC1 via sequestration to so-called “HIC1 bodies” (fig. 1C). In the following section we will discuss how inactivation of HIC1 and hence induction of the HIC1-repressed targets might lead to cancer.
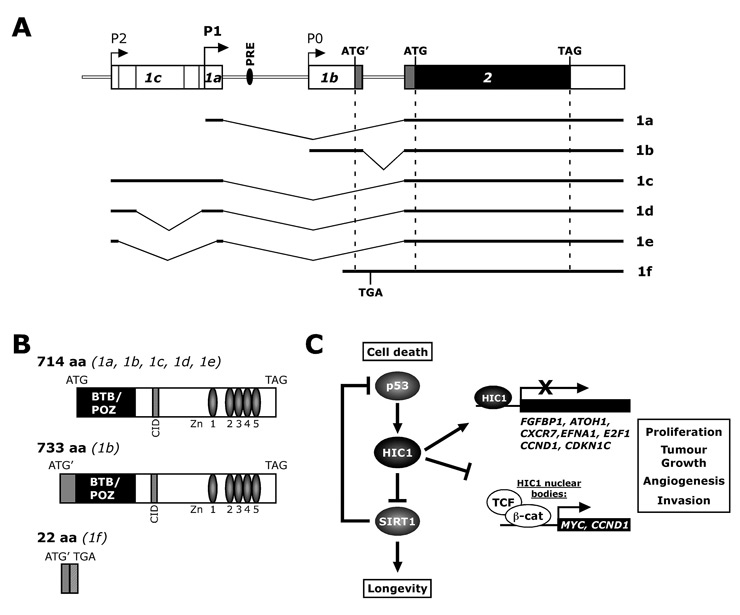
Figure 1
Gene structure of human HIC1. (A) Schema of the human HIC1 gene: HIC1 transcription can initiate at three separate promoters (P0, P1 and P2) that give rise to three alternative first exons 1a, 1b and 1c, followed by a second coding exon, which contains the 3’ untranslated region. Exons 1a and 1c are noncoding and associated with the major GC-rich promoters P1 and P2 respectively, whereas exon 1b is partially coding and associated with the P0 TATA box promoter. Exon 1b contains an ATG’ codon that is in frame with the ATG initiation codon located in exon 2. Additional alternative splice events in exon 1c yield two new transcripts named 1d and 1e, both transcribed from promoter P2. Translation of the unspliced transcript 1f starts at ATG’ within exon 1b, but contains a TGA stop codon in the unspliced intron sequence. Possible translational starts (ATG; ATG’, alternative start codon of 1b and f) and stops (TAG; TGA, stop codon of unspliced 1f) are indicated. Black and grey colouring represents translated exons. PRE, p53 responsive element. (B) Functional protein domains of HIC1 and possible HIC1 isoforms. Splicing variants leading to the respective protein isoform are indicated. Transcript 1b could encode an alternative protein containing additional 19 amino acids (12 derived from exon 1b and 7 from 5’ sequences in exon 2) at the N-terminus. The unspliced transcript 1f might result in a 22 amino acid polypeptide due to a premature stop codon in the intron. BTB/BOZ, broad complex, tramtrack and bric à brac/poxviruses and zinc finger repression domain; CID, consensus motif of the CtBP-interacting domain; Zn, zinc fingers. (C) Putative HIC1 tumour suppressor pathways. Please see text for details. FGFBP1, Fibroblast growth factor binding protein; ATOH1, Atonal Homolog 1; CXCR7, chemokine (C-X-C motif) receptor 7; EFNA1, Ephrin-A1; CCND1, Cyclin D1; CDKN1C, cyclin-dependent kinase inhibitor 1C/p57KIP2.
The silent mating type information regulation 2 homolog 1 (SIRT1) deacetylase was the first transcriptional target of HIC1 to be identified [14]. SIRT1 belongs to the type III NAD+-dependent histone/protein deacetylases family and is involved in regulating cellular senescence and longevity. In line with this concept, enhanced SIRT1 expression has been found in different human cancers [15]. An important, non-histone substrate of SIRT1 is the p53 tumour suppressor. Deacetylation of the p53 transcription factor attenuates its ability to activate downstream targets involved in regulation of apoptosis and/or proliferation. HIC1 directly interacts with the SIRT1 protein, forming a transcriptional repression complex which then binds and represses the SIRT1 promoter [14]. Further, the authors suggested a model where under normal physiological conditions HIC1 represses SIRT1transcription and therefore inhibits p53 deacetylation. Indeed, acetylation is indispensable for p53 activation, to control growth arrest and apoptosis in response to stress such as DNA damage. Since p53 is a positive transcriptional regulator of HIC1, activated p53 would induce HIC1 expression, which in turn represses SIRT1, thus representing a positive feedback loop. In tumour cells where HIC1 is inactivated, SIRT1 levels may increase, leading to deacetylation and therefore inactivation of p53 activity and allowing cells to bypass apoptosis and survive DNA damage. However, a recent publication did not find a correlation between HIC1 and SIRT1 expression in primary diffuse large B-cell lymphomas, arguing against direct regulation of SIRT1 by functional HIC1 [16].
Briones et al. described an HIC1 binding site in the fibroblast growth factor binding protein (FGF-BP;FGFBP1) promoter that is necessary to mediate repression of FGF-BP [17]. FGF-BP has been shown to enhance FGF-mediated biochemical and biological events specifically during blood vessel growth. For example, constitutive expression of FGF-BP resulted in highly angiogenic tumours in xenograft tumour assays. Additionally, blocking FGF-BP inhibited proliferation of prostate cancer cells [18]. In conclusion, inactivation of HIC1 in tumour cells may allow expression of FGF-BP, resulting in an increase in angiogenesis and/or proliferation.
Hic1 was recently described as a novel tumour suppressor in a murine medulloblastoma model [19], where it cooperates with the Patched (Ptch) 1tumour suppressor. Ptch1
+/–
/Hic1
+/– heterozygous knockout mice showed a markedly increased incidence of medulloblastoma as compared to Ptch1
+/–heterozygous mice. Moreover, it was found that the proneural transcription factor Atonal Homolog 1 (Atoh1) is directly suppressed by Hic1. Atoh1 is essential for cerebellar growth and development. The authors suggested a model where inactivation of Hic1 leads to untimely Atoh1 expression which then might support the malignant growth of medulloblastoma.
Another direct HIC1 target is the scavenger chemokine receptor 7 (CXCR7). It was identified as one of several down-regulated genes in a genome-wide expression profiling in HIC1-deficient U2OS osteosarcoma cells transduced with an adenoviral HIC1-expression vector. Further analyses, including confirmatory real-time PCR and promoter analyses using luciferase assays and chromatin immunoprecipitation, showed that HIC1 forms a complex with the co-repressor CtBP and binds to HIC1-responsive elements in the CXCR7 promoter to repress its expression [20]. It is therefore conceivable that loss-of-HIC1-function in tumour cells leads to increased CXCR7 expression. Indeed, CXCR7 is highly expressed in primary human breast and lung cancer [21]. In line with this concept, suppression of CXCR7 by RNAi reduces in vivo tumour growth in nude mice [22]. Taken together, by regulating CXCR7 expression, HIC1 may be involved in regulation of the chemokine cross-talk between tumour cells and the surrounding stroma.
There is yet another mechanism whereby HIC1 has been implicated in epithelial malignancy: HIC1 has recently been shown to be a direct transcriptional repressor of the gene encoding ephrin-A1 (EFNA1) [23]. Similarly to the above studies, HIC1 has been shown to bind the ephrin-A1 promoter directly and repress transcription. Ephrin-A1 functions as a cell surface ligand for Eph receptors, which are a subfamily of receptor tyrosine kinases. Bidirectional ephrin/Eph-signalling has been
implicated in many aspects of malignancy, such as tumour growth, invasion, metastasis and angiogenesis (reviewed in [24]). Consequently, in breast cancer cells restoration of HIC1 function reduced tumour growth in vivo, which could partly be reversed by ectopic expression of ephrin-A1.
In addition to being a transcriptional target of E2F1 [11], HIC1 in turn suppresses E2F-responsive genes, such as E2F1, by recruiting corepressors of the SWI/SNF family to the promoters [25]. This forms another feedback loop finetuning the actions of the E2F family members. E2Fs are crucial in regulating cell cycle progression, and inactivation of HIC1 leads to undue activation of E2F signalling, thus favouring cell cycle progression and tumour growth.
Lastly, HIC1is involved in regulation of the T-cell-specific transcription factor 4 (TCF4), although by a different inhibitory mechanism. TCF4belongs to the T-cell receptor/lymphoid enhancer binding factor (TCR/LEF) protein family, and as its relatives it functions as a nuclear effector of the Wnt signalling pathway. TCF4interacts with β-catenin in active Wnt signalling and co-activates downstream target genes. This activity is important during normal development, but its deregulation plays a pivotal role in cancer progression, as evidenced by the frequent disruption of the negative Wnt regulator APC (adenomatous polyposis coli) in colorectal cancer. Earlier it was shown that HIC1 sequestered C-terminal binding proteins (CtBPs) to nuclear dot-like structures called HIC1-bodies [8]. Valenta et al. [26] have now found that HIC1 also sequestered TCF4 as well as TCF4 bound β-catenin via CtBP to HIC1-bodies and thereby attenuated Wnt signalling. This may suppress tumour formation, since TCF4 and β-catenin are prevented from activating TCF-responsive genes possibly involved in tumour development, such as c-Myc (MYC) or Cyclin D1 (CCND1). Interestingly, Cyclin D1, a positive cell cycle regulator that is frequently amplified in tumours, was also identified as a direct HIC1-repressed gene [20, 27]. The same study revealed the cell cycle inhibitor P57KIP2 (CDKN1C) as a novel HIC1 target. This apparent contradiction of HIC1 repressing a cell cycle accelerator, Cyclin D1 and inhibitor, P57KIP2,may be explained by the observation that low levels of P57KIP2are able to promote cyclin/CDK complex formation and thus cell cycle progression [28].
HIC1 inactivation in solid cancers
HIC1 promoter hypermethylation was found in a wide variety of solid cancers, for example in breast, brain, liver, colorectal, cervical and lung tumours (see table 1 for a detailed list). Importantly, while few studies have tried to link HIC1 promoter hypermethylation to HIC1 mRNA expression, it is generally assumed that hypermethylation of the HIC1promoter region leads to silencing of HIC1 gene expression [7, 29]. On the other hand, Fujii et al. found that in normal breast ductal tissue one HIC1allele is often hypermethylated, whereas both alleles were hypermethylated in the major portion of primary breast cancer tissue [30]. In line with this concept, two studies found increasing HIC1 promoter hypermethylation from normal liver tissue, to precancerous liver tissue showing chronic hepatitis or cirrhosis, to primary hepatocellular carcinoma [29, 31]. Overall HIC1 expression levels decreased during the development of cancer, but hypermethylation did not correlate significantly with HIC1 expression levels in hepatocellular carcinoma cells or the corresponding noncancerous tissue, indicating that HIC1 inhibitory mechanisms other than hypermethylation contribute to the low HIC1expression.
In addition, HIC1 promoter hypermethylation was found in normal brain tissue of children [32], in adult brain [33] and in prostate epithelium [34], but since HIC1 expression was not measured it cannot be concluded that these normal tissues are predisposed to tumour development due to low HIC1 expression. In summary, these findings indicate that low HIC1 levels contribute to cancer development, but also that inhibitory mechanisms other than
hypermethylation of the HIC1 promoter exist, e.g. mutations/inactivation of the positive HIC1 regulator p53, or aberrant expression of not yet identified HIC1inhibitors.
|
Table 1
HIC1 promoter hypermethylation in primary solid tumours. |
|
Tissue
|
Technique
|
Hyper-methylation
|
Expression
|
Reference
|
| Colorectal cancer |
MSED |
38–80% |
n.d. |
[44] |
| Colorectal cancer |
MSP |
62% |
n.d. |
[45] |
| Colorectal cancer |
ML |
52% |
n.d. |
[46] |
| Non-small cell lung cancer |
MSED |
31–33% |
n.d. |
[47] |
| Hepatocellular carcinoma* |
MSED |
38–90% |
+ |
[29] |
| Breast cancer* |
MSED |
67% |
+ |
[30] |
| Breast cancer* |
MSP |
43–64% |
n.d. |
[48] |
| Cervical cancer |
MSP |
9–30% |
n.d. |
[49] |
| Cervical cancer |
MSP |
59% |
n.d. |
[50] |
| Cervical adenocarcinoma |
MSP, BGS |
32–64% |
n.d. |
[51] |
| Ovarian carcinoma |
MSP, COBRA |
11–39% |
n.d. |
[52] |
| Ovarian carcinoma |
MSP |
34% |
n.d. |
[53] |
| Bladder carcinomas |
MSP |
0% |
n.d. |
[54] |
| Prostate cancer |
MSED |
96% |
n.d. |
[34] |
| Renal tumours* |
MSED |
50–74% |
n.d. |
[55] |
| Glioblastoma* |
MSED |
60% |
n.d. |
[56] |
| Glioma* |
ML |
100% |
n.d. |
[33] |
| Medulloblastoma* |
MSP |
75–90% |
+ |
[57] |
| Ependymonas* |
MSP, BGS |
83% |
+ |
[58] |
| Germ cell tumour |
MSP, BGS |
32% |
n.d. |
[59] |
| Paediatric neoplasms* |
MSP |
0–100% |
CL |
[60] |
| Gastric tumours* |
MSED |
15–45% |
n.d. |
[61] |
| * = including normal tissue; CL = cell lines only; n.d. = not done; BGS = bisulphite genomic sequencing; BiPS = bisulphite treatment/PCR-single strand conformation polymorphism; COBRA = combined bisulphite restriction analysis; ML = MethyLight analysis; MSED = methylation sensitive enzymatic digestion; MSP = methylation specific PCR. |
HIC1 in normal and malignant haematopoiesis
A possible role for HIC1 in leukaemogenesis is suggested by the fact that the distal arm of chromosome 17p is often altered in advanced stages of chronic myeloid leukaemia (CML) or acute lymphoid leukaemia (ALL). In a first publication, Issa et al. investigated whether HIC1 is hypermethylated in a panel of different haematopoietic disorders. Using restriction enzyme methylation analysis of five NotI sites within the HIC1 5’ untranslated region (promoter P0), they found that normal haematopoietic cells, such as peripheral blood leukocytes, CD34- or CD34+ cells, were not hypermethylated. In contrast, HIC1 promoter methylation was seen in ALL, in chronic-phase CML (CML-CP) as well as in primary non-Hodgkin’s lymphoma (NHL) [35]. Moreover, they found increased HIC1 methylation in ALL patients who relapsed after a chemotherapy-induced complete remission, as well as in CML in blast crisis (CML-BC) as compared to CML-CP. These observations indicate that HIC1 promoter hypermethylation may be a late event in ALL and CML [35].
In the most common type of NHL, diffuse large B-cell lymphoma (DLBCL), Stocklein et al. [16] identified HIC1 as a novel tumour suppressor telomeric to p53. In most cases hypermethylation of one HIC1 allele was accompanied by deletion of the second allele, resulting in HIC1
–/– tumours. Moreover, DLBCL patients with complete inactivation of both p53 and HIC1 genes presented a worse clinical course than patients with inactivation of p53 alone. This functional cooperation of the two proteins in tumour development is supported by data from Hic1
+/–
/p53
+/– double heterozygous mice, which show a higher incidence of osteosarcoma and lymphoma than Hic1 altered mice [36].
Only 10% of primary acute myeloid leukaemia (AML) showed methylation of the HIC1 P0 promoter at diagnosis, suggesting that HIC1 promoter hypermethylation is an infrequent event in this disease [35]. Ekmekci et al. did not find HIC1 promoter hypermethylation in a second cohort of AML [37], and recently low levels (12.5%) of HIC1 promoter hypermethylation were found in myelodysplastic syndromes or secondary AML from MDS [38]. Other studies found higher levels of HIC1 hypermethylation in MDS (32%, [39]) and in 107 AML patients (overall 51%, [40]). It is worth noting that the second study explicitly excluded AML of the M3 subtype (acute promyelocytic leukaemia, APL). A different study found hypermethylation of exon 2 (referred to intron 2 and exon 3 in their publication) in 10 of 12 AML samples. Importantly, no correlation between HIC1 mRNA levels and methylation of exon 2 was found in AML or bone marrow from healthy donors [41]. Taken together, there are a significant number of AML cases that do not exhibit HIC1 promoter hypermethylation. In agreement with those results, we found that the P0 and P1 HIC1 promoters are not hypermethylated in HL60 promyelocytic cells as measured by bisulphite genomic sequencing [42]. Nevertheless, primary AML samples showed significantly lower HIC1 expression than granulocytes, and HIC1 was upregulated in different models of myeloid differentiation. Hence the reasons behind the low expression of HIC1 in AML remain unclear. A finding of note was a 10-fold higher induction of HIC1 in the t(15;17)-positive APL cell line NB4 than in HL60 or U937 myeloid leukaemic cells (fig. 2A and [42]) upon all-trans retinoic acid (ATRA)-induced granulocytic differentiation. To rule out the possibility that enhanced HIC1 induction in NB4 cells is due to a higher basal level of promoter methylation – ATRA effects include inhibition of methyltransferases – we analysed the HIC1 promoter in NB4 cells. As seen with HL60 cells, no hypermethylation of the HIC1 P0 promoter was found. As a control we used U2OS osteosarcoma cells that showed dense hypermethylation of P0 (fig. 2B). We only investigated the P0 promoter because tumours that do not express HIC1 most often experience P0 promoter hypermethylation [43]. These findings indicate that other inhibitory mechanisms than hypermethylation must be active in NB4 cells. Since the major molecular difference between HL60 and NB4 cells is the expression of promyelocyte leukaemia – retinoic acid receptor α (PML-RARα) fusion protein in NB4 cells, and because the HIC1 promoter is not hypermethylated in these cells and
because HIC1 induction upon ATRA treatment is higher than in PML-RARa negative cells, we speculate that PML-RARa may repress HIC1 transcription. In line with this concept, we identified a putative ATRA responsive element upstream of exon 2 (fig. 2C). Ongoing research aims to elucidate the role of RARa/PML-RARa in HIC1 regulation. Obviously, a PML-RARα HIC1 repression model would only explain low HIC1 levels in APL and not in other AML subtypes, and therefore further studies are needed to identify novel HIC1 inhibitory pathways in AML.
Conclusions
HIC1 is a central transcriptional regulator of key genes controlling cell growth as well as cell death in response to DNA damage. Alteration of the HIC1pathway may lead to abnormal cell proliferation and stress responses contributing to a cancerous phenotype. Accordingly, HIC1 is frequently hypermethylated in a variety of solid tumours and leukaemias, making it an interesting therapeutic target for hypomethylating agents such as decitabine. Moreover, low HIC1 expression in the absence of HIC1 promoter hypermethylation, as seen in certain leukaemias, points to additional inhibitory pathways that may serve as starting points for new therapeutic approaches.
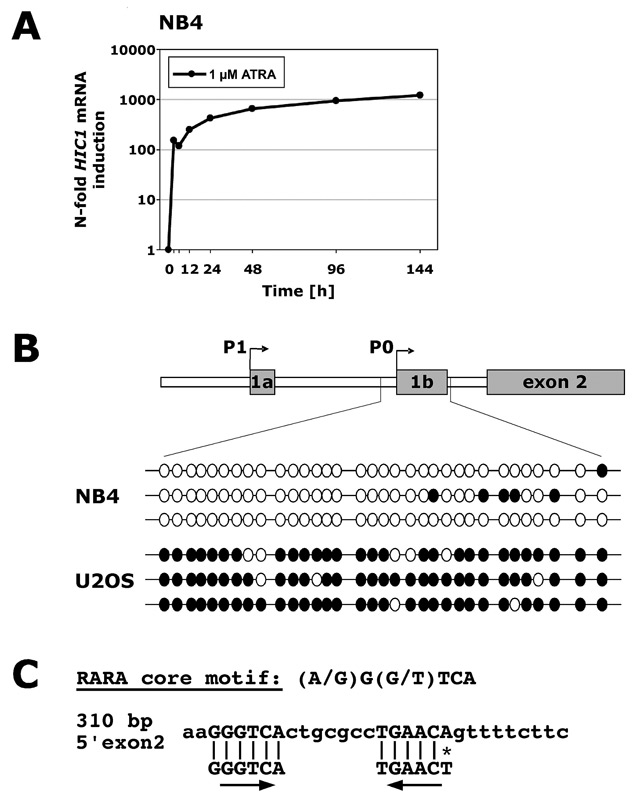
Figure 2
HIC1 regulation in acute promyelocytic leukaemia (APL). (A) NB4 APL cells were differentiated towards granulocytes in vitro for times indicated using 1 µM all-trans retinoic acid (ATRA). Successful differentiation was confirmed by morphology and FACS analysis of CD11b surface expression. Total RNA was extracted and levels of HIC1 mRNA were assessed using real-time quantitative RT-PCR. mRNA levels are expressed as n-fold changes in regulation compared to untreated cells using HMBS mRNA expression as a reference gene. (B) Bisulphite sequencing of NB4 APL and U2OS osteosarcoma cells. Top: Genomic organisation of the HIC1 promoter region highlighting the analysed CpG island. Methylation was assessed by sequencing of three individual clones derived by PCR on bisulphite-treated genomic DNA. Black circles represent methylated CpG sites, white circles unmethylated CpG sites, and each horizontal line indicates an individual allele. (C) Putative retinoic acid receptor binding element in the HIC1 promoter. Homologies to a region 5’ of exon 2 as well as orientation of the two core motifs are indicated.
Correspondence to:
Mario P. Tschan, PhD
Experimental Oncology/Haematology
MEM E829
Department of Clinical Research
University of Bern
Murtenstrasse 35
CH-3010 Bern
Switzerland
mtschan@dkf.unibe.ch
References
1 Carter MG, Johns MA, Zeng X, Zhou L, Zink MC, Mankowski JL, Donovan DM, Baylin SB. Mice deficient in the candidate tumor suppressor gene Hic1 exhibit developmental defects of structures affected in the Miller-Dieker syndrome. Hum Mol Genet. 2000;9:413–9.
2 Guerardel C, Deltour S, Pinte S, Monte D, Begue A, Godwin AK, Leprince D. Identification in the human candidate tumor suppressor gene HIC-1 of a new major alternative TATA-less promoter positively regulated by p53. J Biol Chem. 2001;276:3078–89.
3 Pinte S, Guerardel C, Deltour-Balerdi S, Godwin AK, Leprince D. Identification of a second G-C-rich promoter conserved in the human, murine and rat tumor suppressor genes HIC1. Oncogene. 2004;23:4023–31.
4 Mondal AM, Chinnadurai S, Datta K, Chauhan SS, Sinha S, Chattopadhyay P. Identification and functional characterization of a novel unspliced transcript variant of HIC-1 in human cancer cells exposed to adverse growth conditions. Cancer Res. 2006;66:10466–77.
5 Deltour S, Guerardel C, Leprince D. Recruitment of SMRT/N-CoR-mSin3A-HDAC-repressing complexes is not a general mechanism for BTB/POZ transcriptional repressors: the case of HIC-1 and gammaFBP-B. Proc Natl Acad Sci. USA 1999;96:14831–6.
6 Pinte S, Stankovic-Valentin N, Deltour S, Rood BR, Guerardel C, Leprince D. The tumor suppressor gene HIC1 (hypermethylated in cancer 1) is a sequence-specific transcriptional repressor: definition of its consensus binding sequence and analysis of its DNA binding and repressive properties. J Biol Chem. 2004;279:38313–24.
7 Wales MM, Biel MA, el Deiry W, Nelkin BD, Issa JP, Cavenee WK, et al. p53 activates expression of HIC-1, a new candidate tumour suppressor gene on 17p13.3. Nat Med. 1995;1:570–7.
8 Deltour S, Pinte S, Guerardel C, Wasylyk B, Leprince D. The human candidate tumor suppressor gene HIC1 recruits CtBP through a degenerate GLDLSKK motif. Mol Cell Biol. 2002;22:4890–901.
9 Stankovic-Valentin N, Verger A, Deltour-Balerdi S, Quinlan KG, Crossley M, Leprince D. A L225A substitution in the human tumour suppressor HIC1 abolishes its interaction with the corepressor CtBP. FEBS J. 2006;273:2879–90.
10 Britschgi C, Rizzi M, Grob TJ, Tschan MP, Hugli B, Reddy VA, et al. Identification of the p53 family-responsive element in the promoter region of the tumor suppressor gene hypermethylated in cancer 1. Oncogene. 2006;25:2030–9.
11 Jenal M, Trinh E, Britschgi C, Britschgi A, Roh V, Vorburger SA, et al. The tumor suppressor gene hypermethylated in cancer 1 is transcriptionally regulated by E2F1. Mol Cancer Res. 2009;7:916–22.
12 Putzer BM. E2F1 death pathways as targets for cancer therapy. J Cell Mol Med. 2007;11:239–51.
13 Fleuriel C, Touka M, Boulay G, Guerardel C, Rood BR, Leprince D. HIC1 (Hypermethylated in Cancer 1) epigenetic silencing in tumors. Int J Biochem Cell Biol. 2009;41:26–33.
14 Chen WY, Wang DH, Yen RC, Luo J, Gu W, Baylin SB. Tumor suppressor HIC1 directly regulates SIRT1 to modulate p53-dependent DNA-damage responses. Cell. 2005;123:437–48.
15 Huffman DM, Grizzle WE, Bamman MM, Kim JS, Eltoum IA, Elgavish A, Nagy TR. SIRT1 is significantly elevated in mouse and human prostate cancer. Cancer Res. 2007;67:6612–8.
16 Stocklein H, Smardova J, Macak J, Katzenberger T, Holler S, Wessendorf S, et al. Detailed mapping of chromosome 17p deletions reveals HIC1 as a novel tumor suppressor gene candidate telomeric to TP53 in diffuse large B-cell lymphoma. Oncogene. 2008;27:2613–25.
17 Briones VR, Chen S, Riegel AT, Lechleider RJ. Mechanism of fibroblast growth factor-binding protein 1 repression by TGF-beta. Biochem Biophys Res Commun. 2006;345:595–601.
18 Aigner A, Renneberg H, Bojunga J, Apel J, Nelson PS, Czubayko F. Ribozyme-targeting of a secreted FGF-binding protein (FGF-BP) inhibits proliferation of prostate cancer cells in vitro and in vivo. Oncogene. 2002;21:5733–42.
19 Briggs KJ, Corcoran-Schwartz IM, Zhang W, Harcke T, Devereux WL, Baylin SB, et al. Cooperation between the Hic1 and Ptch1 tumor suppressors in medulloblastoma. Genes Dev. 2008;22:770–85.
20 Van Rechem C, Rood BR, Touka M, Pinte S, Jenal M, Guerardel C, et al. Scavenger chemokine (CXC motif) receptor 7 (CXCR7) is a direct target gene of HIC1 (hypermethylated in cancer 1). J Biol Chem. 2009;284:20927–35.
21 Miao Z, Luker, K.E., Summers, B.C., Berahovich, R., Bhojani, M.S., Rehemtulla, A., Kleer, C.G., Essner, J.J., Nasevicius, A., Luker, G.D., Howard, M.C., Schall, T.J. (2007) CXCR7 (RDC1) promotes breast and lung tumor growth in vivo and is expressed on tumor-associated vasculature. Proc Natl Acad Sci U S A 104, 15735-40.
22 Burns JM, Summers BC, Wang Y, Melikian A, Berahovich R, Miao Z, et al. A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J Exp Med. 2006;203:2201–13.
23 Zhang W, Zeng X, Briggs KJ, Beaty R, Simons B, Chiu Yen RW, et al. A potential tumor suppressor role for Hic1 in breast cancer through transcriptional repression of ephrin-A1. Oncogene. 2010;29:2467–76.
24 Pasquale EB. Eph receptors and ephrins in cancer: bidirectional signalling and beyond. Nat Rev Cancer. 2010;10:165–80.
25 Zhang B, Chambers KJ, Leprince D, Faller DV, Wang S. Requirement for chromatin-remodeling complex in novel tumor suppressor HIC1-mediated transcriptional repression and growth control. Oncogene. 2009;28:651–61.
26 Valenta T, Lukas J, Doubravska L, Fafilek B, Korinek V. HIC1 attenuates Wnt signaling by recruitment of TCF-4 and beta-catenin to the nuclear bodies. EMBO J. 2006;25:2326–37.
27 Van Rechem C, Boulay G, Pinte S, Stankovic-Valentin N, Guerardel C, Leprince D. Differential regulation of HIC1 target genes by CtBP and NuRD, via an Acetylation/SUMOylation switch, in quiescent versus proliferating cells. Mol Cell Biol.
28 Pateras IS, Apostolopoulou K, Niforou K, Kotsinas A, Gorgoulis VG. p57KIP2: “Kip”ing the cell under control. Mol Cancer Res. 2009;7:1902–19.
29 Kanai Y, Hui AM, Sun L, Ushijima S, Sakamoto M, Tsuda H, Hirohashi S. DNA hypermethylation at the D17S5 locus and reduced HIC-1 mRNA expression are associated with hepatocarcinogenesis. Hepatology. 1999;29:703–9.
30 Fujii H, Biel MA, Zhou W, Weitzman SA, Baylin SB, Gabrielson E. Methylation of the HIC-1 candidate tumor suppressor gene in human breast cancer. Oncogene. 1998;16:2159–64.
31 Nishida N, Nagasaka T, Nishimura T, Ikai I, Boland CR, Goel A. Aberrant methylation of multiple tumor suppressor genes in aging liver, chronic hepatitis, and hepatocellular carcinoma. Hepatology. 2008;47:908–18.
32 Rood BR, Zhang H, Weitman DM, Cogen PH. Hypermethylation of HIC-1 and 17p allelic loss in medulloblastoma. Cancer Res. 2002;62:3794–7.
33 Uhlmann K, Rohde K, Zeller C, Szymas J, Vogel S, Marczinek K, et al. Distinct methylation profiles of glioma subtypes. Int J Cancer. 2003;106:52–9.
34 Morton RA Jr, Watkins JJ, Bova GS, Wales MM, Baylin SB, Isaacs WB. Hypermethylation of chromosome 17P locus D17S5 in human prostate tissue. J Urol. 1996;156:512–6.
35 Issa JP, Zehnbauer BA, Kaufmann SH, Biel MA, Baylin SB. HIC1 hypermethylation is a late event in hematopoietic neoplasms. Cancer Res. 1997;57:1678–81.
36 Chen W, Cooper TK, Zahnow CA, Overholtzer M, Zhao Z, Ladanyi M, et al. Epigenetic and genetic loss of Hic1 function accentuates the role of p53 in tumorigenesis. Cancer Cell 2004;6:387–98.
37 Ekmekci CG, Gutierrez MI, Siraj AK, Ozbek U, Bhatia K. Aberrant methylation of multiple tumor suppressor genes in acute myeloid leukemia. Am J Hematol. 2004;77:233–40.
38 Grovdal M, Karimi M, Khan R, Aggerholm A, Antunovic P, Astermark J, et al. Maintenance treatment with azacytidine for patients with high-risk myelodysplastic syndromes (MDS) or acute myeloid leukaemia following MDS in complete remission after induction chemotherapy. Br J Haematol. 2010.
39 Aggerholm A, Holm MS, Guldberg P, Olesen LH, Hokland P. Promoter hypermethylation of p15INK4B, HIC1, CDH1, and ER is frequent in myelodysplastic syndrome and predicts poor prognosis in early-stage patients. Eur J Haematol. 2006;76:23–32.
40 Deneberg S, Grovdal M, Karimi M, Jansson M, Nahi H, Corbacioglu A, et al. Gene-specific and global methylation patterns predict outcome in patients with acute myeloid leukemia. Leukemia. 2010;24:932–41.
41 Melki JR, Vincent PC, Clark SJ. Cancer-specific region of hypermethylation identified within the HIC1 putative tumour suppressor gene in acute myeloid leukaemia. Leukemia. 1999;13:877–3.
42 Britschgi C, Jenal M, Rizzi M, Mueller BU, Torbett BE, Andres AC, et al. HIC1 tumour suppressor gene is suppressed in acute myeloid leukaemia and induced during granulocytic differentiation. Br J Haematol. 2008;141:179–87.
43 Chen WY, Zeng X, Carter MG, Morrell CN, Chiu Yen RW, Esteller M, et al. Heterozygous disruption of Hic1 predisposes mice to a gender-dependent spectrum of malignant tumors. Nat Genet. 2003;33:197–202.
44 Ahuja N, Mohan AL, Li Q, Stolker JM, Herman JG, Hamilton SR, et al. Association between CpG island methylation and microsatellite instability in colorectal cancer. Cancer Res. 1997;57:3370–4.
45 Goel A, Nagasaka T, Arnold CN, Inoue T, Hamilton C, Niedzwiecki D, et al. The CpG island methylator phenotype and chromosomal instability are inversely correlated in sporadic colorectal cancer. Gastroenterology. 2007;132:127–38.
46 Nosho K, Kure S, Irahara N, Shima K, Baba Y, Spiegelman D, et al. A prospective cohort study shows unique epigenetic, genetic, and prognostic features of synchronous colorectal cancers. Gastroenterology. 2009;137:1609–20 e1-3.
47 Eguchi K, Kanai Y, Kobayashi K, Hirohashi S. DNA hypermethylation at the D17S5 locus in non-small cell lung cancers: its association with smoking history. Cancer Res. 1997;57:4913–5.
48 Parrella P, Scintu M, Prencipe M, Poeta ML, Gallo AP, Rabitti C, et al. HIC1 promoter methylation and 17p13.3 allelic loss in invasive ductal carcinoma of the breast. Cancer Lett. 2005;222:75–81.
49 Gustafson KS, Furth EE, Heitjan DF, Fansler ZB, Clark DP. DNA methylation profiling of cervical squamous intraepithelial lesions using liquid-based cytology specimens: an approach that utilizes receiver-operating characteristic analysis. Cancer. 2004;102:259–68.
50 Kim JH, Choi YD, Lee JS, Lee JH, Nam JH, Choi C. Assessment of DNA methylation for the detection of cervical neoplasia in liquid-based cytology specimens. Gynecol Oncol. 2010;116:99–104.
51 Dong SM, Kim HS, Rha SH, Sidransky D. Promoter hypermethylation of multiple genes in carcinoma of the uterine cervix. Clin Cancer Res. 2001;7:1982–6.
52 Teodoridis JM, Hall J, Marsh S, Kannall HD, Smyth C, Curto J, et al. CpG island methylation of DNA damage response genes in advanced ovarian cancer. Cancer Res. 2005;65:8961–7.
53 Feng Q, Deftereos G, Hawes SE, Stern JE, Willner JB, Swisher EM, et al. DNA hypermethylation, Her-2/neu overexpression and p53 mutations in ovarian carcinoma. Gynecol Oncol. 2008;111:320–9.
54 Kunze E, Wendt M, Schlott T. Promoter hypermethylation of the 14-3-3 sigma, SYK and CAGE-1 genes is related to the various phenotypes of urinary bladder carcinomas and associated with progression of transitional cell carcinomas. Int J Mol Med. 2006;18:547–57.
55 Makos M, Nelkin BD, Reiter RE, Gnarra JR, Brooks J, Isaacs W, et al. Regional DNA hypermethylation at D17S5 precedes 17p structural changes in the progression of renal tumors. Cancer Res. 1993;53:2719–22.
56 Li Q, Jedlicka A, Ahuja N, Gibbons MC, Baylin SB, Burger PC, Issa JP. Concordant methylation of the ER and N33 genes in glioblastoma multiforme. Oncogene. 1998;16:3197–202.
57 Zawlik I, Zakrzewska M, Witusik M, Golanska E, Kulczycka-Wojdala D, Szybka M, et al. KCTD11 expression in medulloblastoma is lower than in adult cerebellum and higher than in neural stem cells. Cancer Genet Cytogenet. 2006;170:24–8.
58 Waha A, Koch A, Hartmann W, Mack H, Schramm J, Sorensen N, et al. Analysis of HIC-1 methylation and transcription in human ependymomas. Int J Cancer. 2004;110:542–9.
59 Koul S, Houldsworth J, Mansukhani MM, Donadio A, McKiernan JM, Reuter VE, et al. Characteristic promoter hypermethylation signatures in male germ cell tumors. Mol Cancer. 2002;1:8.
60 Rathi A, Virmani AK, Harada K, Timmons CF, Miyajima K, Hay RJ, et al. Aberrant methylation of the HIC1 promoter is a frequent event in specific pediatric neoplasms. Clin Cancer Res. 2003;9:3674–8.
61 Kanai Y, Ushijima S, Ochiai A, Eguchi K, Hui A, Hirohashi S. DNA hypermethylation at the D17S5 locus is associated with gastric carcinogenesis. Cancer Lett. 1998;122:135–41.