DOI: https://doi.org/10.4414/smw.2010.13108
Autoimmunity is the result of the end of tolerance to a self-antigen. It occurs under physiological conditions as a participant in homeostasis without causing any damage (i.e., natural autoimmunity). However, under pathological conditions, the loss of self-tolerance induces the development of autoimmune diseases (AD) such as rheumatoid arthritis (RA), systemic lupus erythematosus (SLE) or multiple sclerosis (MS). The pathogenesis of these disorders, associated with genetic, epigenetic and environmental factors [1], involves both cognate and innate immunity. While in these diseases the autoantigens are inconstantly characterised, the involvement of autoreactive B and T lymphocytes and the role of pro-inflammatory partners are better defined and have been extensively studied. The exact aetiology of AD is unknown and their treatment relies on anti-inflammatory and immunosuppressive drugs, with no specificity for the pathogenic mechanisms of the disease.
Although clinically distinct, all AD share some similarities in their pathogenesis (deregulation of T-cell and B-cell activation, production of autoantibodies, etc…) and involve the production of cytokines and chemokines, which are important protein mediators that specifically regulate the inflammatory response, tissue damage and repair mechanisms [2]. Cytokines can be distinguished as “pro-inflammatory” such as TNF-α, IL-1β, IL-17 or IL-6, and “anti-inflammatory” such as IL-4, IL-10 or IL-13. They both act on innate or cognate phases of the immune processes, playing a role in AD’s induction, regulation and amplification [3].
The increasing physiopathological knowledge of the regulatory role of these soluble and/or membrane-bound messengers has lead to the development of anti-cytokine molecules, like monoclonal antibodies (mAbs) which have subsequently been approved for clinical use [4]. Although the efficacy of mAbs passive immunotherapy in treating AD is largely recognised, some drawbacks such as high costs and primary or secondary unresponsiveness remain. The role of these treatments in the development of secondary diseases such as infections (and possibly even cancer) is a matter of concern as well [5]. These concerns justify the efforts to develop alternative cytokine-targeting strategies. Among them is active immunotherapy.
Active immunotherapy is based on the simple and well-established vaccination principle and on the natural capacity of immune system to mount a physiological response to a given antigen. For vaccination against self-proteins, such as anti-cytokine active immunotherapy (ACAI), the aim is to obtain high titers of neutralising anti-cytokine antibodies with no associated cellular response. Indeed, accumulation of auto-reactive T cells in the site of inflammation can induce irreversible tissue damage, with potentially life-threatening consequences [6]. For this purpose, efficient induction of the humoral response involves breaking B tolerance and induction of specific T helper cells.
Regarding ACAI, induction of anti-cytokine autoantibodies production is not difficult. Recently, autoantibodies against cytokines, such as TNF-α, IL-2, IL-8 or VEGF without associated neutralising capacity in vivo, have been observed in healthy individuals, demonstrating their physiological production [7]. Presence of autoantibodies against cytokines has been described in several chronic diseases, without highlighting any regulatory or worsening role of these proteins [8, 9]. One of the major points of ACAI is to obtain anti-cytokine autoantibodies with a high neutralising capacity, so that they could be used for therapeutic applications. The mechanism of action results from the blockade of the activity of the pathogenic cytokine by neutralising antibodies. In addition, the binding of any antibody from the desired polyclonal response against the targeted cytokine could lead to the formation of immune complexes that would then be cleared: it may lower the cytokine level to a point where a clinical benefit is achieved.
Although this review will only cover vaccination in autoimmune diseases, this technique is also used in non-autoimmune diseases such as tumours. In cancer, a major target is a pro-angiogenic growth factor, VEGF. Recently, a vaccine composed of VEGF coupled to keyhole limpet hemocyanine (KLH) reduced angiogenesis in murine tumour models. The VEGF kinoid (VEGF-K) induced the production of high titers of neutralising anti-VEGF antibodies, inhibiting the binding of VEGF to its receptors [10]. A recent clinical trial proved the efficacy of vaccination against angiotensin II in mild to moderate hypertension [11].
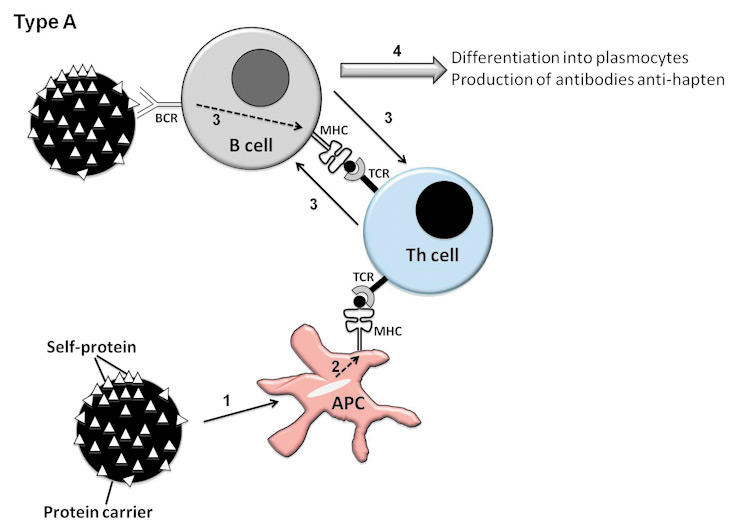
Two different methods (types of vaccination) can be used to circumvent the Th tolerance to autoantigens and induce anti-cytokines autoantibodies. Type I vaccination consists of using the protein of interest to engineer a vaccine. Two approaches have to be differentiated [12]. Although it is conceivable to use xenogeneic antigens, the development of vaccination in autoimmune diseases has used self-antigens. The first description of such a vaccination is that by Dalum et al., who introduced foreign immunodominant T-helper epitopes in the native structure of the cytokine of interest (type I B vaccination) (fig. 1). This method proved effective in collagen-induced arthritis (CIA) in mice, a model of RA [13] and in a mouse model of asthma [14]. Alternatively, the self-protein can be modified by linking it to a foreign carrier protein in order to provide adequate help to activate autoreactive B-cells (type I A) (fig. 1). Vaccination with such a hetero-complex, made of a biologically inactive cytokine and carrier protein, induces a carrier-specific Th-cell proliferation helping the self-cytokine-specific B cells to produce autoantibodies. Repetitive antigens coupled to a carrier protein induce a strong activation stimulus which is able to overcome the B-cell tolerance. The carrier can be synthetic virus like particles (VLPs) [15], KLH (16) or ovalbumin (OVA) [17]. The T-cell response is supported by the carrier molecule, and a B-cell response is directed versus epitopes present on the targeted protein.
Another approach to induce autoantibodies against cytokines is by using a plasmid DNA vaccination (type II vaccination). This alternative vaccination method can provide an immunogenic antigen processed via class I and II major histocompatibility complex (MHC); capable of stimulating immunological memory, and containing an adjuvant [18]. Induction of protective therapy against AD could be achieved by targeted DNA vaccines encoding pro-inflammatory cytokines, such as TNF-α in CIA [19] or experimental autoimmune encephalomyelitis (EAE) [20]. The presence of hypomethylated immunostimulatory sequences (CpG) in such a vaccine may explain the ability of certain plasmid to serve as an adjuvant via a TLR9 pathway [21]. The cells transfected by the plasmid produce the self-protein. The endogenous B epitopes produced are recognised by BCR of autoreactive B cells. A direct transfection of the auto-reactive B cells, in which the CpG sequences are linked to the TLR9, leads to the B cells activation. To explain the breaking of the B tolerance, another hypothesis is the activation of Th cells specific for epitopes of the endogenous protein [21].
Rheumatoid arthritis (RA) is the most frequent autoimmune chronic inflammatory rheumatism, primarily affecting the synovial membrane of multiple joints. Although its aetiology is still unknown, it is now acknowledged that during the inflammatory process of arthritis there are three key mediators, the proinflammatory cytokines TNF-α, IL-1β and IL-6 [22]. The dramatic efficacy of TNF-α blockade by monoclonal antibodies has generated interest in developing alternative strategies for antagonising TNF-α, such as gene therapy by electrotransfer [23], and siRNA [24]. At the present time, passive immunotherapy with monoclonal antibodies or type II receptor of TNF-α is the only strategy to target this cytokine in RA and other TNF-α-dependent diseases (spondylarthropathies, Crohn’s disease, psoriasis).
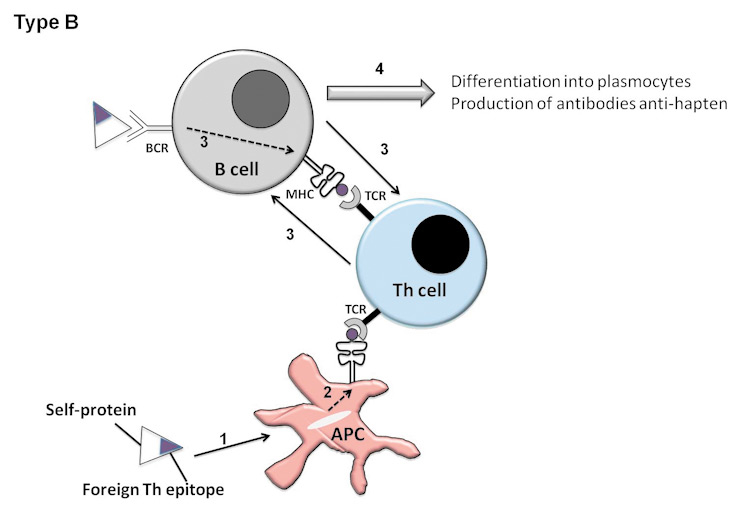
Figure 1
Hypothesis for the rupture of tolerance to self-antigen in strategies of type I vaccination, consisting of using a modified self-protein. Type A (Coupling of a self-protein to a carrier protein). The following steps occur subsequently: 1: The APC phagocytes the carrier protein (black circle). 2: The APC presents carrier epitopes to Th cells through their MHCII. 3: B cells specific of self-protein (small white triangles) are activated through T-B cooperation. 4: Differentiation of B cells into plasmocytes producing anti-hapten antibodies Type B (Introduction of a foreign Th epitope into the native protein of interest). The following steps occur subsequently: 1: The APC phagocytes the foreign Th epitope. 2: the APC present the foreign epitope to the Th cells. 3: B cells specific of self-protein are activated through T-B cooperation. 4: Differentiation of B cells into plasmocytes producing anti-hapten antibodies.
Several strategies to block TNF-α by coupling this cytokine with a carrier protein have been developed. A vaccine composed of biologically inactive human TNF-α coupled to KLH, (TNF Kinoid, TNF-K) [25], induced production of high titers of neutralising anti-human TNF-α (hTNF-α) antibodies in different strains of mice [26]. In 1991, Keffer et al.developed an experimental model of spontaneous arthritis based on the over-expression of hTNF-α in transgenic mice [27]. Vaccination with TNF-K of these hTNF-α transgenic mice, which develop severe arthritis from 8–10 weeks of age [28], protected them against clinical and histological arthritis in short and long-term experiments [26, 29], even when the vaccination was performed after onset of arthritis [16]. Indeed, TNF-K-vaccinated mice firstly showed a clinical and histological improvement and then, several weeks post TNF-K primo-injection, a clinical worsening paralleled by a decrease of anti-hTNF-α antibodies titer (bell curve). Both clinical worsening and anti-hTNF-α antibodies titer’s decline were reversed by a maintenance dose of TNF-K. Additionally, it has been demonstrated that no B-cell memory response to hTNF-α was induced by TNF-K immunisation. Indeed, injection of native hTNF-α after immunisation by TNF-K does not induce the production of neutralising anti-TNF-α autoantibodies [16]. Based on these data, confirming the reversibility and repeatability of TNF-K vaccination, a phase II clinical trial led by Néovacs (Paris, France) was approved in RA and is ongoing at present [16].
The first immunisation against TNF-α was that described by Dalum et al. in 1999 [13]. They demonstrated that symptoms of cachexia and CIA can be ameliorated by vaccination with recombinant Th epitope modified murine TNF-α without inducing a significant T-cell response against TNF-α. Amelioration of CIA was also demonstrated using a murine TNF-α vaccine, constituted of multiple copies of TNF-α peptides coupled to VLP [30, 31]. Vaccination with a plasmid encoding heterologous TNF-α induced anti-TNF-α antibodies and prevented CIA in mice via cross-reaction [19]. In another mouse model of experimental arthritis, adjuvant arthritis, injection of naked DNA encoding autologous TNF-α suppressed the disease [32].
The pro-inflammatory cytokine IL-1β is another important mediator of inflammation and a major cause of tissue damage in RA. Intra-articular injections of IL-1β induce synovial inflammation, leucocytes synovial infiltration and proteoglycans depletions [33]. The therapeutic administration of the recombinant IL-1 receptor antagonist (IL-1Ra, anakinra) is capable to reduce clinical symptoms of disease. Yet, it suffers from several drawbacks including the need for frequent high-dose administration [34]. To counteract these drawbacks, computer-designed IL-1β peptides located in the potential sites of interaction between the cytokine and its receptor have been synthesised. They were then coupled to the KLH and tested in CIA [35]. More recently, a vaccine composed of IL-1α or IL-1β chemically cross-linked to VLP of the bacteriophage Qβ elicited a rapid and long-lasting autoantibody response. In the CIA model, both vaccines strongly protected mice from inflammation and degradation of bone and cartilage [34].
After the discovery of the T-cell cytokine interleukin-17 (IL-17) in the synovium of RA patients, research on the role of IL-17 in the arthritis process were developed [36]. The efficacy of IL-17-specific monoclonal antibodies in ameliorating inflammatory diseases in animal models yielded the development of active immunotherapy targeting IL-17. Vaccination with VLP conjugated with recombinant IL-17 (IL-17-VLP) induced high titers of neutralising anti-IL-17 antibodies and reduced scores of arthritis in a CIA model [15].
With the final aim being application in RA, other anti-cytokine vaccines have been designed and tested in several experimental models (table 1). One of them is an anti-IL-6 vaccine constituted of immunogenic IL-6 analogues. This vaccine was effective to protect mice from CIA, while in LPS-induced systemic inflammation model, it significantly raised TNF-α levels [37]. An anti-MIF vaccination, with MIF/tetanus toxoid DNA vaccine, inhibited the development of arthritis in collagen antibody-induced arthritis (CAIA) and in IL-1Ra deficient mice [38]. In rats, vaccination with plasmid DNA encoding IL-27 p28 given at the onset or during the course of adjuvant-induced arthritis (AIA), protected from AIA compared to empty plasmid vaccinated animals [39]. Other active immuno-therapies have been demonstrated to be efficient in AIA by Youssef et al. These therapies are directed against macrophage-inflammatory protein-1α (MIP-1α) and protein-1β (MIP-1β), monocyte chemoattractant protein 1 (MCP-1) and RANTES (CCL-5) in AIA [40, 41].
| Table 1Applications of anti-cytokine vaccinations in rheumatic diseases. | ||||
| Cytokine target | Product | Disease | Animal species | References |
| mTNFα | mTNFα / Th foreign epitopes | CIA, Cachexia | Mouse | [13] |
| hTNFα | hTNFα / KLH | arthritis | Hum. Transg. Mouse | [26] |
| rTNFα | rTNFα plasmide | AIA | Rat | [32] |
| mTNFα | hTNFα plasmide | CIA | Mouse (Kumming) | [19] |
| mTNFα | TNFα-peptide / VLPbiot. | CIA | Mouse | [30] |
| mTNFα | mTNFα-TNFa peptide / VLPbiot. | CIA | Mouse | [31] |
| mIL1-β | mIL-1α peptides / KLH | CIA | Mouse | [35] |
| mIL1 (α and β) | mIL-1α and mIL-1β / VLP Qβ | CIA | Mouse | [34] |
| mIL-6 | mIL6 analogues | CIA | Mouse | [37] |
| mIL-12 | mIL-12p40 / Ova/PADRE / KLH/BSA | Leishmaniosis | Mouse | [52] |
| mIL-12 p40 / PADRE | Atherosclerosis | Mouse LDL-R–/– | [49] | |
| mIL-17 | mIL-17A / VLP Qβ | CIA, CAIA | Mouse | [15] |
| mIL-18 | mIL-18 plasmide | Lupus | Mouse lpr | [48] |
| mIL-27 | mIL-27 p28 plasmide | AIA | Rat | [39] |
| mMIF | MIF | CAIA, IL-1Ra–/– | Mouse | [38] |
| rMIP-1α/1β | MIP-1α and MIP-1β plasmids | AIA | Rat | [41] |
| rMCP-1 | MCP-1 plasmid | AIA | Rat | [41] |
| rRANTES | RANTES plasmid | AIA | Rat | [41] |
Multiple sclerosis (MS) is the most common inflammatory demyelinating disease of the central nervous system (CNS). The animal model for human disease is the experimental autoimmune encephalomyelitis (EAE). EAE and MS share circulating leukocytes penetration of the blood brain barrier and myelin damage, impaired nerve conduction and paralysis [42]. The disease initiation and progression are mediated by antigen-specific T cells. The CD4+ T cells can be divided into different subsets characterised by their cytokine profile. The Th2 subset produces IL-4, IL-5 and IL-13. The Th1 cells produce large amount of IFN-γ and TNF-α. These cytokines seem to have a predominant role in initiation and progression of the inflammatory process in several autoimmune diseases, including EAE [42]. However, according to recent studies, many autoimmune processes previously described as Th1 dependent could be actually due to Th17 cells. EAE, for instance, is mediated by activated effector/memory Th17 cells. These cells cross the blood-brain barrier, and release cytokines leading to infiltration of other immune cell types that eventually exert a demyelinating activity [15]. Both anti-IL-17 and anti-TNF-α vaccines were designed to treat EAE. Two anti-IL-17 vaccines have been developed. In the first, IL-17 was coupled to VLP. As for CIAC (see above), this vaccine was found to be efficacious even in EAE [15]. In the second case, IL-17A dimmers were conjugated to OVA. Different subtypes of Il-17 exist but only two are implied during inflammation: IL-17A and F. IL-17A was chosen because its affinity for the IL-17 receptor (IL-17R) is better than for IL-17F. IL-17A has also been shown to be essential for the IL-17R activation. This vaccine, like the first one, was efficacious in treating EAE [17]. Furthermore, a TNF-α-naked DNA vaccine enhanced the production of TNF-α-specific antibody titer and conferred EAE resistance in EAE rats models [43].
Recruitment of leukocytes in the brain in MS depends on chemokines and chemotactic cytokines. The anti-C-C chemokine-based immunotherapy was efficacious in mice EAE. The mice were passively immunised by rabbit anti-mouse polyclonal Abs against macrophage-inflammatory protein-1α (MIP-1α) [44]. To target the function of key pro-inflammatory chemokines, Youssef et al. applied the naked DNA vaccination. For this purpose, Lewis rats were immunised with M1P-α and MCP-1 vaccines. It was demonstrated that these vaccines prevent EAE even when the disease was induced two months after administration of naked DNA vaccines [20, 45].
SLE is a chronic inflammatory disease with pan-systemic involvement. The predominant role of type I IFN in the development of the disease is based on both detection of high IFN-α serum levels in SLE patients and on a relatively benign disease course in mice with defective type I IFN receptors [46]. Mathian et al. developed a SLE-like flare model in which the expression of IFN-α from a recombinant adenovirus induced early expression disease manifestations. Recently, a vaccine composed of IFN-α coupled to KLH, IFN-K, was tested in this experimental model. Immunisation of mice with IFN-K induced transient neutralising antibodies and allowed a significant decrease in disease manifestations, including proteinuria, histological renal lesions and death, as long as anti-IFN-α antibodies were present in the sera of immunised mice [47].
Related to the IL-1 cytokine family, and described as an IFN-γ-inducing factor, IL-18 is a cytokine with a strong Th1-promoting activity. In autoimmune and chronic inflammatory disorders, such as SLE, this cytokine has a pro-inflammatory role. MLR/Mp-Tnfrs6lpr (lpr) mouse is a model of SLE. After immunisation with a plasmid DNA encoding IL-18, the mice produced autoantibodies against the cytokine leading to a decrease of spontaneous lymphoproliferation and to a reduction of disease severity and renal damage [48].
Atherosclerosis is an inflammatory disease in which IL-12 favours the development of a pro-atherosclerotic Th-1 cell phenotype. This heterodimeric cytokine is composed of a 35kDa light chain and a 40kDa heavy chain. It is possible to detect IL-12 in human atherosclerotic plaques, and in mice models it has been associated with the initiation as well as the acceleration of atherosclerosis. An IL-12p35 coupled to PADRE vaccine allowed the production of anti-IL-12 antibodies and reduced atherogenesis in LDL-receptor deficient mice, without any change in serum cholesterol level [49].
Active anti-cytokine immunotherapy has also been applied to autoimmune myocarditis. Infection by some viruses, bacteria or protozoa can cause acute heart-muscle inflammation (myocarditis) that can be complicated by dilated cardiomyopathy. In fact, in some susceptible individuals, the infection can be followed by an autoimmune response against heart-muscle myosin, leading to myocardial dilation and heart failure. Many pro-inflammatory cytokines are involved in the development of the disease. These cytokines such as IL-1, IL-6 and TNF-α are mainly produced by dendritic cells (DCs) and macrophages. As Th17 has a major role in the disease, an anti-IL-17 vaccine has been developed and tested in an experimental model of autoimmune myocarditis. Vaccination by IL-17-VLP led to high titers of anti-IL-17 antibodies and to a protection against autoimmune myocarditis [50].
Diabetes mellitus is also an autoimmune disease in which vaccination strategy has been applied by coupling a modified IL-1β to VLP. Indeed, IL-1β is strongly implicated in the pathogenesis of type II diabetes through the destruction of pancreatic islet cells that produce insulin [51]. Recently, in June 2009, Cytos biotechnology started a phase I/II trial in patients with type II diabetes with an anti-IL-1 vaccine. ( http://www.clinicaltrial.gov ).
A large body of evidence confirms that anti-cytokine vaccination has entered the era of being a serious alternative immunotherapy. The main targets are cytokines involved in the chronic inflammatory process. Since the cytokines play a major role in homeostasis, some concerns have to be pointed out, representing important points for further development of this strategy. First is the mid and long-term safety of such an approach; in an initial analysis, the question of the persistence of the anti-cytokine antibodies has to be ascertained. A bell-curve over a reasonable period of time (few weeks or months) comparable to that of passive immunotherapy, should represent a guarantee. In other cases, the innocuity of the anti-cytokine antibodies should be demonstrated. A second major point is the T-cell response to the target cytokine. To prevent immune dysregulation due to the persistent over-expression of a given cytokine it seems in fact mandatory to select methods that do not enhance the cytokine-targeted T-cell response.
The ongoing clinical trials will give some answers about this promising approach of a new class of biologics. To end with, even if these are non-scientific arguments, the easiness of use and the likely reduction of costs of such treatments need to be mentioned.
1 Baranzini SE. The genetics of autoimmune diseases: a networked perspective. Curr Opin Immunol. 2009;21(6):596–605.
2 Delavallee L, Assier E, Denys A, et al. Vaccination with cytokines in autoimmune diseases. Ann Med. 2008;40(5):343–51.
3 Oppenheim JJ, Feldmann M. Introduction to the role of cytokines in innate host defense and adaptative immunity. Cytokine References volume 1: Ligands. Oppenheim JJ, Feldmann M (Eds). Academic Press, London, UK, 3–20 (2000).
4 Krause I, Valesini G, Scrivo R, et al. Autoimmune aspects of cytokine and anticytokine therapies. Am J Med. 2003;115(5):390–7.
5 Bruns A, Nicaise-Roland P, Hayem G, et al. Prospective cohort study of effects of infliximab on rheumatoid factor, anti-cyclic citrullinated peptide antibodies and antinuclear antibodies in patients with long-standing rheumatoid arthritis. Joint Bone Spine. 2009;76(3):248–53.
6 Ferrer I, Boada Rovira M, Sanchez Guerra ML, et al. Neuropathology and pathogenesis of encephalitis following amyloid-beta immunization in Alzheimer’s disease. Brain Pathol. 2004;14(1):11–20.
7 Watanabe M, Uchida K, Nakagaki K, et al. Anti-cytokine autoantibodies are ubiquitous in healthy individuals. FEBS Lett. 2007;581(10):2017–21.
8 Rosenau BJ, Schur PH. Autoantibodies to tumor necrosis factor in patients with rheumatoid arthritis and systemic lupus erythematosus. J Rheumatol. 2009;36(4):753–6.
9 Graudal NA, Svenson M, Tarp U, et al. Autoantibodies against interleukin 1alpha in rheumatoid arthritis: association with long term radiographic outcome. Ann Rheum Dis. 2002;61(7):598–602.
10 Rad FH, Le Buanec H, Paturance S, et al. VEGF kinoid vaccine, a therapeutic approach against tumor angiogenesis and metastases. Proc Natl Acad Sci USA. 2007;104(8):2837–42.
11 Tissot AC, Maurer P, Nussberger J, et al. Effect of immunisation against angiotensin II with CYT006-AngQb on ambulatory blood pressure: a double-blind, randomised, placebo-controlled phase IIa study. Lancet. 2008;371(9615):821–7.
12 Delavallee L, Assier E, Semerano L, et al. Emerging applications of anticytokine vaccines. Expert Rev Vaccines. 2008;7(10):1507–17.
13 Dalum I, Butler DM, Jensen MR, et al. Therapeutic antibodies elicited by immunization against TNF-alpha. Nat Biotechnol. 1999;17(7):666–9.
14 Tan GH, Wang CC, Huang FY, et al. Active immunotherapy of allergic asthma with a recombinant human interleukin-5 protein as vaccine in a murine model. Chin Med J. (Engl). 2007;120(17):1517–22.
15 Rohn TA, Jennings GT, Hernandez M, et al. Vaccination against IL-17 suppresses autoimmune arthritis and encephalomyelitis. Eur J Immunol. 2006;36(11):2857–67.
16 Delavallee L, Semerano L, Assier E, et al. Active immunization to tumor necrosis factor-alpha is effective in treating chronic established inflammatory disease: a long-term study in a transgenic model of arthritis. Arthritis Res Ther. 2009;11(6):R195.
17 Uyttenhove C, Van Snick J. Development of an anti-IL-17A auto-vaccine that prevents experimental auto-immune encephalomyelitis. Eur J Immunol. 2006;36(11):2868–74.
18 Tighe H, Corr M, Roman M, et al. Gene vaccination: plasmid DNA is more than just a blueprint. Immunol Today. 1998;19(2):89–97.
19 Shen Y, Chen J, Zhang X, et al. Human TNF-alpha gene vaccination prevents collagen-induced arthritis in mice. Int Immunopharmacol. 2007;7(9):1140–9.
20 Youssef S, Wildbaum G, Maor G, et al. Long-lasting protective immunity to experimental autoimmune encephalomyelitis following vaccination with naked DNA encoding C-C chemokines. J Immunol. 1998;161(8):3870–9.
21 He X, Tsang TC, Zhang T, et al. Antigen epitope-expressing cytokines for DNA immunization. Vaccine. 2005;23(16):1966–72.
22 Dieude P. Rheumatic diseases: environment and genetics. Joint Bone Spine. 2009;76(6):602–7.
23 Bloquel C, Denys A, Boissier MC, et al. Intra-articular electrotransfer of plasmid encoding soluble TNF receptor variants in normal and arthritic mice. J Gene Med. 2007.
24 Khoury M, Escriou V, Courties G, et al. Efficient suppression of murine arthritis by combined anticytokine small interfering RNA lipoplexes. Arthritis Rheum. 2008;58(8):2356–67.
25 Bizzini B, Achour A. “Kinoids”: the basis for anticytokine immunization and their use in HIV infection. Cell Mol Biol (Noisy-le-grand). 1995;41(3):351–6.
26 Le Buanec H, Delavallee L, Bessis N, et al. TNFalpha kinoid vaccination-induced neutralizing antibodies to TNFalpha protect mice from autologous TNFalpha-driven chronic and acute inflammation. Proc Natl Acad Sci USA. 2006;103(51):19442–7.
27 Keffer J, Probert L, Cazlaris H, et al. Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. Embo J. 1991;10(13):4025–31.
28 Hayward MD, Jones BK, Saparov A, et al. An extensive phenotypic characterization of the hTNFalpha transgenic mice. BMC Physiol. 2007;7:13.
29 Delavallee L, Le Buanec H, Bessis N, et al. Early and long-lasting protection from arthritis in tumour necrosis factor alpha (TNFalpha) transgenic mice vaccinated against TNFalpha. Ann Rheum Dis. 2008;67(9):1332–8.
30 Chackerian B, Lowy DR, Schiller JT. Conjugation of a self-antigen to papillomavirus-like particles allows for efficient induction of protective autoantibodies. J Clin Invest. 2001;108(3):415–23.
31 Spohn G, Guler R, Johansen P, et al. A virus-like particle-based vaccine selectively targeting soluble TNF-alpha protects from arthritis without inducing reactivation of latent tuberculosis. J Immunol. 2007;178(11):7450–7.
32 Wildbaum G, Youssef S, Karin N. A targeted DNA vaccine augments the natural immune response to self TNF-alpha and suppresses ongoing adjuvant arthritis. J Immunol. 2000;165(10):5860–6.
33 van de Loo AA, van den Berg WB. Effects of murine recombinant interleukin 1 on synovial joints in mice: measurement of patellar cartilage metabolism and joint inflammation. Ann Rheum Dis. 1990;49(4):238–45.
34 Spohn G, Keller I, Beck M, et al. Active immunization with IL-1 displayed on virus-like particles protects from autoimmune arthritis. Eur J Immunol. 2008;38(3):877–87.
35 Bertin-Maghit SM, Capini CJ, Bessis N, et al. Improvement of collagen-induced arthritis by active immunization against murine IL-1beta peptides designed by molecular modelling. Vaccine. 2005;23(33):4228–35.
36 Boissier MC, Assier E, Biton J, et al. Regulatory T cells (Treg) in rheumatoid arthritis. Joint Bone Spine. 2009;76(1):10–4.
37 Galle P, Jensen L, Andersson C, et al. Vaccination with IL-6 analogues induces autoantibodies to IL-6 and influences experimentally induced inflammation. Int Immunopharmacol. 2007;7(13):1704–13.
38 Onodera S, Ohshima S, Tohyama H, et al. A novel DNA vaccine targeting macrophage migration inhibitory factor protects joints from inflammation and destruction in murine models of arthritis. Arthritis Rheum. 2007;56(2):521–30.
39 Goldberg R, Wildbaum G, Zohar Y, et al. Suppression of ongoing adjuvant-induced arthritis by neutralizing the function of the p28 subunit of IL-27. J Immunol. 2004;173(2):1171–8.
40 Barnes DA, Tse J, Kaufhold M, et al. Polyclonal antibody directed against human RANTES ameliorates disease in the Lewis rat adjuvant-induced arthritis model. J Clin Invest. 1998;101(12):2910–9.
41 Youssef S, Maor G, Wildbaum G, et al. C-C chemokine-encoding DNA vaccines enhance breakdown of tolerance to their gene products and treat ongoing adjuvant arthritis. J Clin Invest. 2000;106(3):361–71.
42 Goldberg R, Zohar Y, Wildbaum G, et al. Suppression of ongoing experimental autoimmune encephalomyelitis by neutralizing the function of the p28 subunit of IL-27. J Immunol. 2004;173(10):6465–71.
43 Wildbaum G, Karin, N. Augmentation of natural immunity to a pro-inflammatory cytokine (TNF-alpha) by targeted DNA vaccine confers long-lasting resistance to experimental autoimmune encephalomyelitis. Gene Ther. 1999;6(6):1128–38.
44 Karpus WJ, Lukacs NW, McRae BL, et al. An important role for the chemokine macrophage inflammatory protein-1 alpha in the pathogenesis of the T cell-mediated autoimmune disease, experimental autoimmune encephalomyelitis. J Immunol. 1995;155(10):5003–10.
45 Youssef S, Wildbaum G, Karin N. Prevention of experimental autoimmune encephalomyelitis by MIP-1alpha and MCP-1 naked DNA vaccines. J Autoimmun. 1999;13(1):21–9.
46 Kunz M, Ibrahim SM. Cytokines and cytokine profiles in human autoimmune diseases and animal models of autoimmunity. Mediators Inflamm. 2009;2009(979258).
47 Zagury D, Le Buanec H, Mathian A, et al. IFNalpha kinoid vaccine-induced neutralizing antibodies prevent clinical manifestations in a lupus flare murine model. Proc Natl Acad Sci USA. 2009;106(13):5294–9.
48 Bossu, P, Neumann, D, Del Giudice E, et al. IL-18 cDNA vaccination protects mice from spontaneous lupus-like autoimmune disease. Proc Natl Acad Sci USA. 2003;100(24):14181–6.
49 Hauer AD, Uyttenhove C, de Vos P, et al. Blockade of interleukin-12 function by protein vaccination attenuates atherosclerosis. Circulation. 2005;112(7):1054–62.
50 Sonderegger I, Rohn TA, Kurrer MO, et al. Neutralization of IL-17 by active vaccination inhibits IL-23-dependent autoimmune myocarditis. Eur J Immunol. 2006;36(11):2849–56.
51 Larsen CM, Faulenbach M, Vaag A, et al. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med. 2007;356(15):1517–26.
52 Uyttenhove C, Arendse B, Stroobant V, et al. Development of an anti-IL-12 p40 auto-vaccine: protection in experimental autoimmune encephalomyelitis at the expense of increased sensitivity to infection. Eur J Immunol. 2004;34(12):3572–81.
Laure Delavallée was the recipient of grants of Arthritis-Fondation Courtin (Paris, France) and the Fondation pour la Recherche Médicale (France). Marie-Christophe Boissier was the recipient of unrestricted grants and/or consultancy fees from Néovacs (Paris, France), Wyeth (France), Pfizer (France), UCB (France).