Multifaceted roles of peroxisome proliferator-activated receptors (PPARs) at the cellular and whole organism levels
DOI: https://doi.org/10.4414/smw.2010.13071
Summary
Chronic disorders, such as obesity, diabetes, inflammation, non-alcoholic fatty liver disease and atherosclerosis, are related to alterations in lipid and glucose metabolism, in which peroxisome proliferator-activated receptors (PPAR)α, PPARβ/δ and PPARγ are involved. These receptors form a subgroup of ligand-activated transcription factors that belong to the nuclear hormone receptor family. This review discusses a selection of novel PPAR functions identified during the last few years. The PPARs regulate processes that are essential for the maintenance of pregnancy and embryonic development. Newly found hepatic functions of PPARα are the mediation of female-specific gene repression and the protection of the liver from oestrogen induced toxicity. PPARα also controls lipid catabolism and is the target of hypolipidaemic drugs, whereas PPARγ controls adipocyte differentiation and regulates lipid storage; it is the target for the insulin sensitising thiazolidinediones used to treat type 2 diabetes. Activation of PPARβ/δ increases lipid catabolism in skeletal muscle, the heart and adipose tissue. In addition, PPARβ/δ ligands prevent weight gain and suppress macrophage derived inflammation. In fact, therapeutic benefits of PPAR ligands have been confirmed in inflammatory and autoimmune diseases, such as encephalomyelitis and inflammatory bowel disease. Furthermore, PPARs promote skin wound repair. PPARα favours skin healing during the inflammatory phase that follows injury, whilst PPARβ/δ enhances keratinocyte survival and migration. Due to their collective functions in skin, PPARs represent a major research target for our understanding of many skin diseases. Taken altogether, these functions suggest that PPARs serve as physiological sensors in different stress situations and remain valuable targets for innovative therapies.
Introduction
Metabolic syndrome is defined as the combination of multiple metabolic disorders, including obesity, dyslipidaemia, glucose intolerance, inflammation and hypertension. The pathophysiology determining this syndrome involves an interaction of key factors, but two of these, insulin resistance and obesity, play major roles. Moreover, low grade inflammation has appeared to be a link between insulin resistance, obesity and type 2 diabetes [1]. In recent years, a number of studies showed that a family of transcription factors, named the peroxisome proliferator-activated receptors (PPARs), improved several of the metabolic abnormalities associated with the metabolic syndrome.
The PPARs are ligand-activated transcription factors which belong to the nuclear hormone receptor superfamily [2–4]. Three isotypes have been identified in lower vertebrates and mammals: PPARα or NR1C1, PPARβ/δ or NR1C2, also called NUC-1 or FAAR, and PPARγ or NR1C3 [5]. These receptors exhibit different tissue distribution and functions and, to some extent, different ligand specificities [6]. Mechanistically, they form heterodimers with the retinoid X receptor (RXR) and activate transcription by binding to a specific DNA element, termed the peroxisome proliferator response element (PPRE), in the regulatory region of a variety of genes encoding proteins that are involved in lipid metabolism and energy balance [7–9]. Binding of agonists within the ligand-binding site of PPARs causes a conformational change promoting binding to transcriptional coactivators. Conversely, binding of antagonists results in a conformation that favours the binding of corepressors [10, 11]. Physiologically, PPAR-RXR heterodimers may bind to PPREs in the absence of a ligand. Although the transcriptional activation depends on the ligand-bound PPAR-RXR, the presence of unliganded PPAR-RXR at a PPRE has effects that vary depending on the promoter context and cell type. For example, for some PPARγ target genes in adipocytes, unliganded PPARγ-RXR heterodimers recruit corepressor complexes, resulting in active repression, whereas for other genes, corepressors are not recruited by unliganded PPARγ-RXR [12]. There are a variety of potential endogenous ligands for the PPARs, such as fatty acids, in particular unsaturated fatty acids, several eicosanoids and metabolites of linoleic acid, which bind to the PPARs with varying affinities, resulting in transcriptional activation of target genes. Synthetic, high-affinity selective ligands for each of the PPARs are also available, as well as dual-specificity ligands that bind with high affinity to these receptors [12].
The predominant role of these receptors, as presently understood, is the transcriptional regulation of enzymes and other proteins involved in energy homeostasis. In addition, crucial roles of PPARα in sexual dimorphism and of PPARβ/δ in tissue repair have also been unveiled [13, 14].
The aim of this review is to summarise new knowledge on the roles of PPARs in the regulation of various physiological and pathophysiological situations, such as energy metabolism in health and diseases, inflammation, sexual dimorphism and tissue repair.
The role of PPARα in sexual dimorphism
During the past decade, a large number of studies concentrated on the main roles of PPARs in energy metabolism and inflammation in response to dietary and endogenous signals. However, their roles extend far beyond these areas. It is common knowledge that the liver, in humans as well as in rodents, plays a crucial role in several gender-dependent metabolic pathways. In fact, the mammalian liver is the centre for sex-specific controls that contribute, in particular, to differences in steroid hormone metabolism and the degradation of xenobiotics. In addition, in both humans and mice, gender affects hepatic inflammatory responses and hepatocellular carcinoma development [15–18]. Many of the 1600 hepatic genes differentially expressed between sexes in the mouse liver are regulated by differences in pituitary growth hormone secretion, which is pulsatile in males and continuous in females. In fact, among the main liver proteins, the cytochrome P450 (CYP) enzymes metabolise diverse steroids, fatty acids and many lipophilic drugs [19, 20]. Though most pronounced in rat and mouse livers, sex differences in CYP expression also occur in humans [21]. The oxysterol 7α-hydroxylase cytochrome P450-b1 (CYP7b1) protein, a prominent member of the CYP family, is highly expressed in male compared to female livers [22–25]. This enzyme has antagonistic actions on sexual hormone production. It negatively regulates the bioavailability of testosterone, the male sexual hormone, and promotes oestrogen receptor (ER) activity by catalysing the clearance of 27-hydroxycholesterol, which functions as a competitive ER antagonist [23, 26]. The activated ER indirectly enhances the expression of CYP7b1, which participates in oestrogen-induced inflammation and hepatotoxicity [27]. Oestrogen-induced hepatotoxicity affects susceptible women using oestrogen-containing oral contraceptives or postmenopausal hormone replacement therapy [27]. Moreover, oestrogens cause intrahepatic cholestasis, the most common hepatic disease during pregnancy, which can result in intrauterine foetal death or spontaneous premature delivery [28]. The cross-talk between ER and PPARα signalling prompted investigation of the role of PPARα in the sex-dependent hepatic gene expression related to steroid metabolism and immunity. Interestingly, sumoylated PPARα mediates female-specific gene repression and protects the liver from oestrogen-induced toxicity in mice [13] (fig. 1A-B). This sex-specific PPARα-dependent gene repression was elucidated using the steroid CYP7b1 gene as a model. The initial sumoylation of the ligand-binding domain of PPARα triggers the interaction of PPARα with the transcription factor GA-binding protein α (GABPα) bound to the Cyp7b1 promoter. This interaction results in the recruitment of histone deacetylase as well as DNA and histone methyltransferases to the promoter. It results in promoter-specific histone deacetylation and methylation, and DNA methylation of a Sp1-binding site adjacent to the GABP binding sites. These events cause the loss of Sp1-stimulated expression of CYP7b1 and thus its downregulation (fig. 1B). Physiologically, this PPARα-dependent gene repression confers protection against oestrogen-induced intrahepatic cholestasis in female mice. In women, oestrogen-induced intrahepatic cholestasis is the most common hepatic disease during pregnancy. The abovementioned findings suggest PPARα as a possible therapeutic target for the prevention or treatment of this disease.
The roles of PPARs in health and disease
The cellular and systemic roles that have been attributed to PPARs extend far beyond the control of hepatic peroxisome proliferation in rodents after which these receptors were initially named [29] (fig. 2). For instance, they have coordinating roles in foetal and early postnatal development [30]. PPARα is highly expressed in the liver, brown adipose tissue, heart, skeletal muscle, kidney and in other organs at lower levels. PPARγ is highly expressed in adipose tissues and is also present in colon and lymphoid organs. PPARβ/δ is expressed ubiquitously, but its levels may vary to a large extent [2].
PPARα
In the liver, PPARα promotes fatty acid oxidation. It is the target for the fibrate class of hypolidaemic drugs such as fenofibrate, clofibrate, and gemfibrozil, which are used for treating hypertriglyceridaemia [7–9]. The role of PPARα in hepatic fatty acid metabolism is especially prominent during fasting (fig. 3A). In fasted PPARα-null mice, its absence is associated with pronounced hepatic steatosis, decreased levels of plasma glucose and ketone bodies, elevated plasma free fatty acid levels and hypothermia [31–33]. These severe metabolic disturbances are the result of the decreased expression of a large number of genes involved in hepatic lipid metabolism, many of which have been identified as direct PPARα target genes [34, 35]. In addition, PPARα is a target of hypothalamic hormone signalling as it plays an important role in the anti-inflammatory action of glucocorticoids [36]. In fact, during fasting, as well as in situations of physical and physiological stress, the hypothalamic ACTH induces the release of glucocorticoids by the adrenal glands, which stimulates the hepatic expression of PPARα (fig. 3A). Moreover, it accumulates according to a daily rhythm with highest levels at the beginning of feeding time [37] (fig. 3A). The availability and production of physiologically relevant ligands of PPARα is central for coordinating its multiple functions in response to the amount and type of food available (fig. 3B) [38, 39].
Interestingly, PPARα is involved in embryonic development and the viability of pups [30]. Although PPARα-null mice are viable, they present an increased risk of maternal abortion and neonatal mortality [40]. In fact, roughly 50% of diabetic PPARα-deficient mice abort between day 12 and 16 of gestation and approximately 80% of their offspring die at birth [40]. Several reasons have been suggested for this increased mortality. Gemfibrozil and clofibrate, two PPARα agonists, down-regulate human chorionic gonadotrophin and up-regulate progesterone secretions in human trophoblast cells, suggesting that a lack of PPARα may be deleterious for the secretion of these hormones, essential for maintaining pregnancy [41]. PPARα is expressed in the placenta in late gestation and all three PPARs (α, β/δ and γ) are expressed in junctional (endocrine functional) and labyrinth (barrier transport) zones of the rat placenta, from day 13–21 of gestation [42, 43]. PPARα has also been observed in the human term placenta, suggesting a potential role in placental fatty acid transfer to meet foetal requirements [43]. These observations show that even if PPARα-null mothers are able to accumulate lipids in the placenta labyrinth, due to the presence of PPARγ, the lack of PPARα may affect foetal-maternal nutrient exchange, which may explain, at least in part, the lethality mentioned above. This placental dysfunction may be more pronounced in diabetes, which increases energy demands. In fact, PPARα-null diabetic mice are hypoinsulinaemic, with a reduced ability to use glucose and lipids as energy sources [40]. Therefore, in addition to hormonal dysfunctions and the high proinflammatory Th1 cytokine levels observed in pregnant PPARα-null diabetic mice and their offspring, the high rate of abortion and mortality rate of pups could also be related to the defect in energy utilisation.
PPARγ
While PPARα controls lipid catabolism and homeostasis in the liver, PPARγ promotes the storage of lipids in adipose tissues. PPARγ is abundantly expressed in adipocytes and plays a pivotal role in adipocyte differentiation [44]. It is a target of the insulin-sensitising thiazolidinediones, a class of drugs which are widely used for the treatment of type 2 diabetes [45] (fig. 2). Despite its relatively low expression levels in healthy liver, PPARγ is critical for the development of hepatic steatosis [46, 47]. The functions of PPARγ extend far beyond fat storage and adipocyte differentiation. For instance, it has a recently recognised role in promoting osteoclast differentiation and bone resorption when activated by rosiglitazone [48]. Its deletion in mouse osteoclast precursors leads to increased bone mass and density and extramedullary haematopoiesis. This phenotype corresponds to the clinical syndrome called osteopetrosis, associated with impaired osteoclast differentiation from haematopoietic stem cells. Conversely, PPARγ down-regulates osteogenesis. In fact, it influences the competition between adipogenic and osteoblastic differentiation of bone marrow progenitors in favour of adipogenesis [49, 50]. Furthermore, all PPARγ-null embryos die at an early developmental stage because of placental defects including impaired vascularisation [51, 52].
PPARβ/δ
This isotype is ubiquitously expressed and is implicated in diverse processes ranging from the regulation of energy homeostasis, thermogenesis, to keratinocyte proliferation and differentiation during wound healing [9]. It stimulates the β-oxidation of fatty acids and reverses cholesterol transport, which lead to the proposal of PPARβ/δ ligands as potential drugs for treating metabolic syndrome [53]. While various physiological functions of PPARβ/δ are still being investigated, roles are already documented in embryo implantation [54], myelination in the brain [55] and osteoclastic bone resorption [56]. Furthermore, its multiple implications in lipid metabolism comprise preadipocyte clonal expansion [57], fatty acid oxidation in muscle [58] and lipoprotein homeostasis [59]. As with PPARα and γ, polyunsaturated fatty acids have a relatively high affinity for PPARβ/δ and a number of eicosanoids, particularly prostacyclin, which is a cyclooxygenase-2 arachidonate metabolite, were shown to activate PPARβ/δ [60].
A key tool for understanding PPAR functions in physiology has been the generation PPAR-null mice. Many PPARβ/δ-null embryos die at an early developmental stage. In fact, PPARβ/δ deletion affects placenta morphogenesis resulting in partial embryonic lethality at embryonic day (E) 9.5 to E10.5 [51]. The trophoblast giant cell layer is most affected, reflecting the involvement of PPARβ/δ in trophoblast cell differentiation toward giant cells [51]. These cells are the primary sites of lipid accumulation in the placenta at an early stage, which correlates with PPARβ/δ-dependent up-regulation of adipose differentiation-related protein (ADRP) expression [51]. In brief, these genetic approaches helped unveil important aspects of the development and functions of the giant cell layer and their impact on placental development.
Limited information is available on PPARβ/δ in the liver, even though it is well expressed in this organ, particularly in endothelial cells and hepatocytes [61, 62]. Data suggest that PPARβ/δ is protective against liver toxicity induced by environmental chemicals, possibly by down-regulating the expression of pro-inflammatory genes [63]. To better understand the function of PPARβ/δ in the liver, the effects of PPARα and PPARβ/δ deletion were compared using whole genome transcriptional profiling. The data obtained provide evidence that PPARβ/δ governs glucose utilization and lipoprotein metabolism and has an important anti-inflammatory role in the liver. In fact, they underscore divergent roles of PPARα and PPARβ/δ in the regulation of gene expression in this organ [64]. Furthermore, in the β-cell line INS-1E, PPARβ/δ protects glucose-stimulated insulin secretion (GSIS) against adverse effects associated with prolonged fatty acid exposure [65]. Finally, a selective activation of PPARβ/δ was shown to promote reverse cholesterol transport [59].
Activation of PPARβ/δ increases lipid catabolism in skeletal muscle, the heart and adipose tissue and improves the serum lipid profile and insulin sensitivity in animal models. It has a crucial role in the distribution of skeletal muscle fibre types [58]. After selective deletion of PPARβ/δ in skeletal muscle, mice exhibit a switch in muscle fibre types toward a lower oxidative capacity that precedes the development of obesity and diabetes, thus demonstrating that PPARβ/δ is instrumental in myocytes for the maintenance of oxidative fibres and that fibre-type switching is likely to be the cause and not the consequence of these metabolic disorders [58]. In addition, PPARβ/δ ligands prevent weight gain and suppress macrophage-derived inflammation [66].
In light of all these reports, PPARs appear to be targets for the treatment of metabolic disorders amongst others. In fact, PPARα and PPARγ are already therapeutic targets for the treatment of hypertriglyceridaemia and insulin resistance, respectively. Evidence is now emerging that PPARβ/δ may also be a potential pharmacological target for the treatment of various metabolic disorders. However, the debate concerning the pro- and anti-cancer properties of this receptor is still open and calls for a scrupulous and thorough assessment of the safety of putative PPARβ/δ drugs.
PPARs and inflammation
In addition to many other cell types, PPARs are expressed in dendritic cells, macrophages, and B and T lymphocytes, suggesting a role in immunity. They are also expressed in epithelial cells, which have an essential function in the mucosal immune response. In line with these observations, many studies have confirmed a therapeutic activity of PPAR ligands in several rodent models of inflammatory and autoimmune diseases [67–69]. For instance, PPARβ/δ ligands have beneficial effects in experimental autoimmune encephalomyelitis (EAE) [70]. PPARα is also found in endothelial cells where it regulates the expression of leukocyte adhesion molecules. In mutant mice, PPARα deficiency shifts the Th1/Th2 balance towards the pro-inflammatory Th1 phenotype [40]. This inflammatory status of PPARα-null mice is exacerbated in the context of diabetic pregnancy and increases the risk of maternal abortion and neonatal mortality (see above). In fact, upon ligand activation, PPARα down-regulates diverse components of the pro-inflammatory response such as chemokines and cytokines by decreasing the expression of the Th1 transcription factor T-bet (T-box expressed in T cells) and increasing the expression of GATA-3 (guanosine adenosine thymidine adenosine 3), known as a positive regulator of Th2 cytokines [71]. PPARα agonists inhibit the transcriptional activity of NF-κB (nuclear factor-κB), AP-1 (activator protein 1), GATA and NFAT (nuclear factor of activated T-cells), which mediate the induction of genes responsible for the development of inflammation and cardiac hypertrophy [72, 73] (fig. 4). The increased severity of inflammatory diseases in PPAR-deficient mice suggests an anti-inflammatory role for either unliganded PPAR or PPAR activated by endogenous ligands [74, 75]. In chronic inflammation, ligand-bound PPARα represses the production of pro-inflammatory IFN-γ and IL-17 by CD4+ T cells and PPARγ ligands modulate the function of dendritic cells to elicit the development of anergic CD4+ T cells [76]. In macrophages, PPARγ ligands repress the expression of a subset of Toll-like receptor (TLR) target genes by a molecular mechanism termed ligand-dependent transrepression [74]. The PPAR ligands also repress the expression of cell adhesion molecules on endothelial cells and the secretion of chemokines by epithelial and other cells, decreasing the recruitment of leukocytes to the site of inflammation [76]. The anti-inflammatory activity of PPAR ligands in mouse models encourages the evaluation of the possibility of PPARs as targets for the treatment of human inflammatory and autoimmune diseases. In fact, PPARα agonists have already been suggested against cardiovascular inflammatory responses. Along the same line of thought, the PPARα agonist fenofibrate is a potent anti-inflammatory drug used in the treatment of patients with rheumatoid arthritis [77]. When used as a lipid-lowering agent in patients with atherosclerosis, fenofibrate induces a decrease in circulating TNF-α, IL-1β and IFN-γ [78]. Finally, the PPARγ agonists thiazolidinediones efficiently normalise skin ho-meostasis when orally administrated to patients suffering from psoriasis, suggesting that their beneficial effects are most likely due to systemic antiinflammatory functions of PPARγ (reviewed in [79]). These reports underscore a promising therapeutic benefit of PPAR ligands in the treatment of inflammatory diseases.
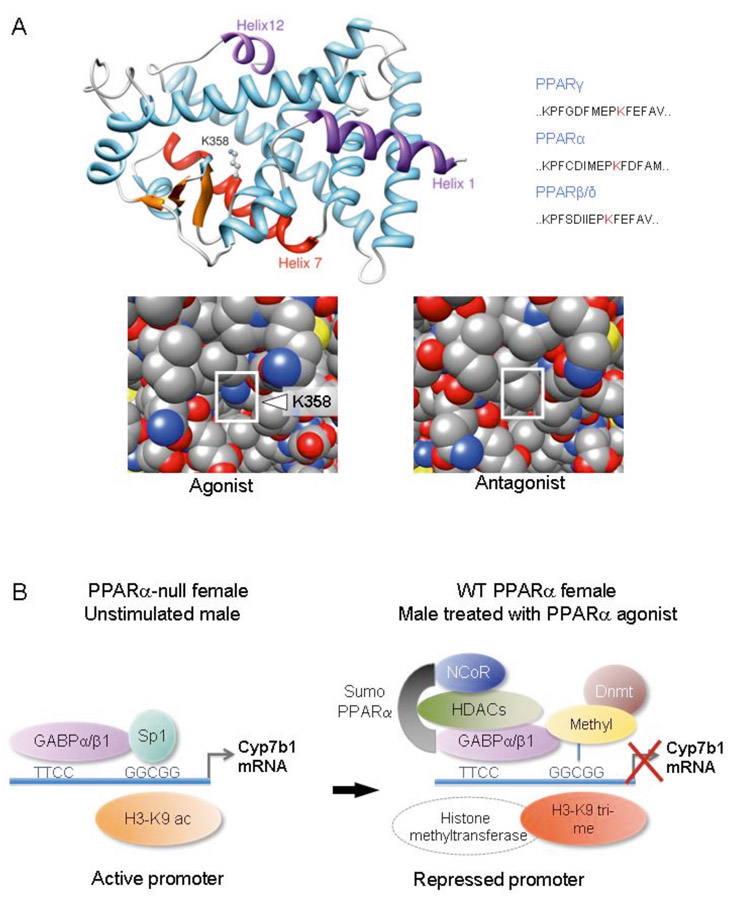
Figure 1
Sumoylation-dependent transrepression by PPARα is sex-dependent.
A.) Model of the ligand-binding domain (LBD) of mouse PPARα in the ligand-activated conformation (upper left panel). Lysine 358 (K358) in Helix 7 of the LBD of PPARα is sumoylated in female hepatocytes and is presented at the surface of the LBD upon agonist binding. This lysine is conserved in the corresponding position of Helix 7 of PPARγ and PPARβ/δ (upper right panel). The change of conformation of Helix 12, which is induced by a PPARα agonist, increases the accessibility of K358 for sumoylation (lower left panel). In contrast, K358 is masked in the presence of an antagonist (lower right panel). The NH-end helix (Helix 1) and COOH-helix (Helix 12) of the ligand binding domain are in pink, Helix 7 with its sumoyalated lysine (K358) in red and the β-sheets in orange.
B.) Model of PPARα-induced repression of Cyp7b1 in females. Sumoylation of PPARα promotes interaction with GABPs and leads to the recruitment of NCoR, HDACs, histone and DNA methyltransferases. These events lead to histone H3 deacetylation, tri-methylation and DNA methylation at the Sp1 binding site, which results in Sp1 displacement and down-regulation of gene expression. After genetic removal of PPARα in female mice the repression complex does not form, which leads to stimulation of Cyp7b1 expression [13].
Cyp7b1, oxysterol 7α-hydroxylase cytochrome P450-b1; HDAC, histone deacetylase; Dnmt, DNA methyltransferase; GABP, GA-binding protein.
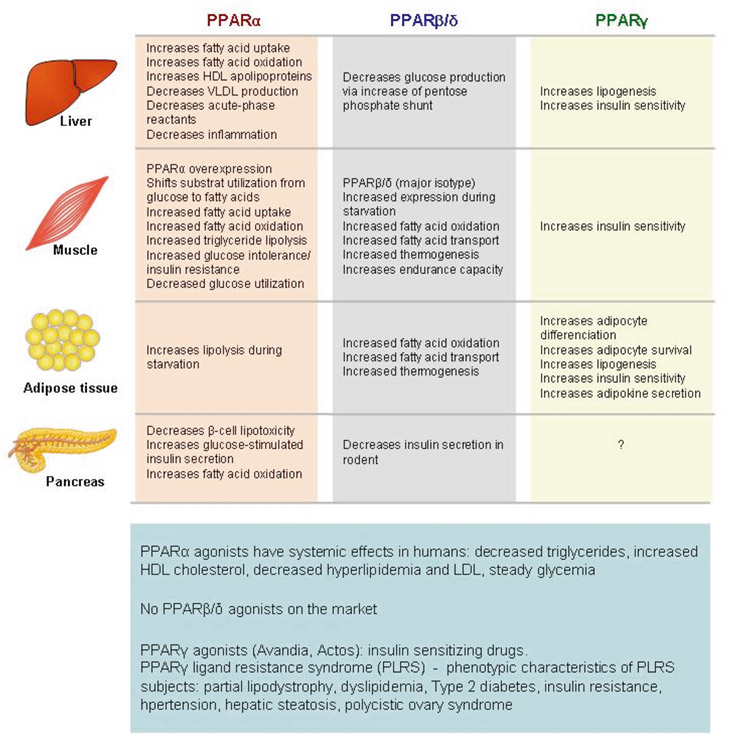
Figure 2
Metabolic functions of PPARs.
Compilation of the known main metabolic functions of PPARα, PPARβ/δ and PPARγ in the liver, muscle, adipose tissue and pancreas.
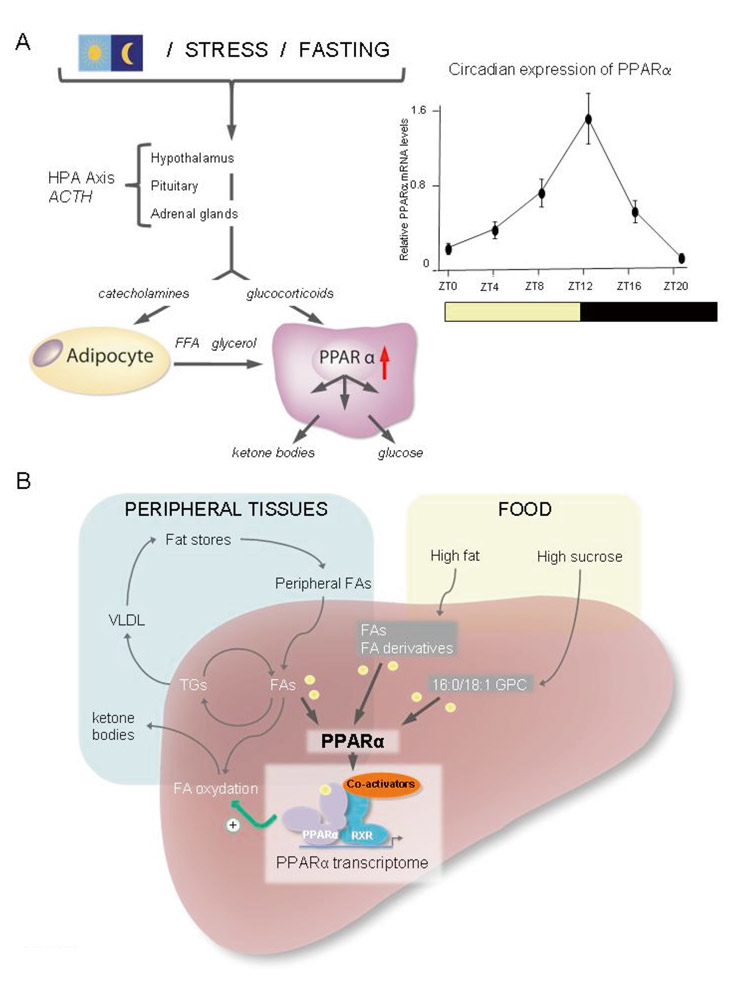
Figure 3
PPARα regulation and activation in the liver.
A.) PPARα expression is stimulated by glucocorticoids (HPA axis; left panel) and follows a circadian rhythm with the highest expression at the end of the light phase (right panel). Lipolysis of triglycerides stored in adipose tissues provides free fatty acids (FFA) and glycerol to the liver, via blood circulation, where they are used for ketone body and glucose production, respectively.
B.) Simplified scheme illustrating the production of ligands activating PPARα. Dietary fatty acids and fatty acid derivatives activate PPARα. On a high sucrose (no fat diet), the production of 16:0/18:1 GPC activates PPARα [38, 39]. Activation of PPARα results in the transcriptional regulation of numerous genes, the so-called PPARα transcriptome, which contributes to maintaining the energy balance in part through the promotion of mitochondrial and peroxisomal beta-oxidation of fatty acids and many other metabolic pathways.
FA, fatty acids; 16:0/18:1 GPC, 1-palmitoyl-2-oleoyl-sn-glycerol-3-phosphocholine; TG, triglycerides; VLDL, very low density lipoprotein.
PPARs are involved in tissue and skin wound repair
The three PPAR isotypes are expressed in rodent and human skin. In addition to their roles in lipid and glucose homeostasis and inflammation (see above), their importance in skin wound healing has emerged more recently [80, 81]. After a skin injury, the healing process requires the covering of the wound bed with a new protective epidermis. This process comprises several phases: inflammation, re-epithelialisation and remodelling of the scar. The initial inflammatory response is followed by the proliferation and migration of keratinocytes, with a parallel dermal repair comprising the activation and proliferation of fibroblasts and angiogenesis. Importantly, PPARα and β/δ are instrumental in this repair process, with each of them playing specific roles [80]. The PPARα isotype is mainly involved in the initial inflammation phase of the pro-cess. It was shown that PPARα-null mice exhibit impaired recruitment of neutrophils and monocytes/macrophages to the wound bed and present a transient delay in healing, which coincides with the inflammatory phase [80]. Transgenic mice with a selective decrease of PPARα activity in keratinocytes only display a similar phenotype, which reveals the importance of keratinocytes in orchestrating the inflammatory process [82]. While the expression of PPARα is transiently up-regulated and is parallel to the inflammatory phase, that of PPARβ/δ in keratinocytes lasts throughout the entire healing process. In agreement with these observations, the completion of skin healing is postponed by two to three days in PPARβ/δ-null mice [80]. PPARβ/δ is an important regulator of keratinocyte survival in the wounded epidermis and is involved in cell adhesion and migration [81] (fig. 5A). In particular, a novel homeostatic control of keratinocyte proliferation and differentiation was recently found, whereby PPARβ/δ regulates the signalling of IL-1 in dermal fibroblasts [83] (fig. 5B). The role of PPARγ appears to be rather minor in keratinocytes, whereas its activity is required for sebaceous gland differentiation. Indeed, activation of PPARγ with troglitazone and prostaglandin-J2 does not affect epidermal maturation in foetal rat skin [84]. However, topical treatments with the PPARγ agonists troglitazone and ciglitazone increase the expression of differentiation markers and promote epidermal barrier recovery in hairless mice [85].
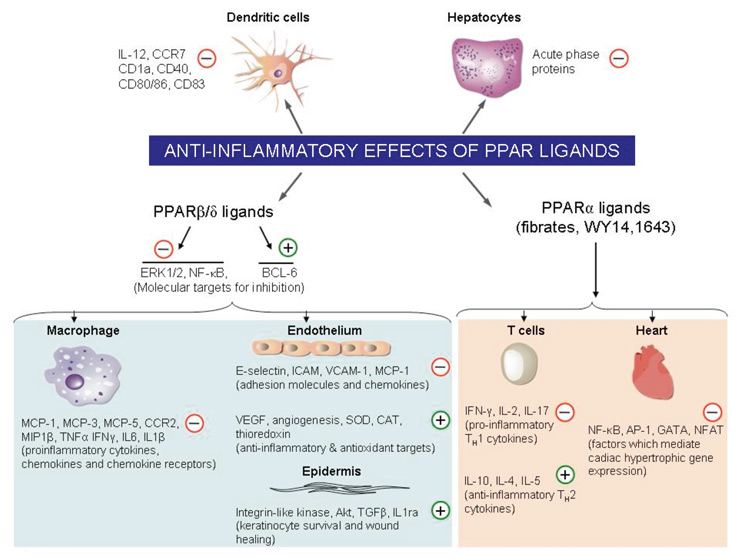
Figure 4
Anti-inflammatory effects of PPAR ligands.
Cellular targets of the anti-inflammatory actions of PPARs activated by both endogenous and synthetic ligands. Ligand-activated PPARs down-regulate or up-regulate diverse components of the inflammatory response. The PPARβ/δ ligands activate the co-repressor BCL-6, inhibit the activity of NF-κB and ERK1/2 and induce anti-inflammatory and antioxidant genes. At the site of inflammation, PPARβ/δ ligands reduce the levels of adhesion molecules (ICAM-1, VCAM-1, E-selectin), pro-inflammatory cytokines (TNFα, IL-6, IL1β, IFNγ), chemokines (MCP-1, -3, -5, MIP1α) and chemokine receptors (CCR2). Furthermore, PPARβ/δ and its endogenous ligands are induced during the inflammatory phase of wound healing and promote keratinocyte migration and survival (Integrin-like kinase and Akt pathways). Antioxidant targets (SOD: superoxide dismutase, CAT: catalase, and thioredoxin) are also induced by PPARβ/δ ligands. The PPARα agonists inhibit transcription factors NF-κB, AP-1, GATA, and NFAT, which mediate the induction of genes responsible for the development of inflammation and cardiac hypertrophy. Moreover, PPARα ligands (fibrates, WY14,643) inhibit the production of Th1 cytokines (IFN-γ, IL-2, IL-17) and induce that of Th2 cytokines (IL-10, IL-4, IL-5) by T cells. Th, T helper cell; (+) positive regulation; (-) negative regulation.
To sum up, we can conclude that PPARα and PPARβ/δ both play important roles in skin wound repair with isotype-specific timing. PPARα favours skin healing via modulation of the inflammatory phase, whilst PPARβ/δ plays an important role in keratinocyte survival and migration. Due to their collective diverse functions in skin biology, PPARs represent major research targets for the understanding and treatment of many skin diseases, such as benign epidermal tumours, papillomas, acne vulgaris and psoriasis.
Conclusion
There are many major cellular and physiological functions regulated by PPARs. At the molecular level, these receptors mainly act through ligand-dependent target gene activation. However, there is increasing evidence for additional modes of action such as transrepression and constitutive activation of the receptors, which also play important roles in PPAR-mediated signalling. Mechanistic studies highlighted promoter-specific regulations occurring through coordinated actions of coregulators and through the integration of extra- and intra-cellular signalling pathways via post-translational modifications.
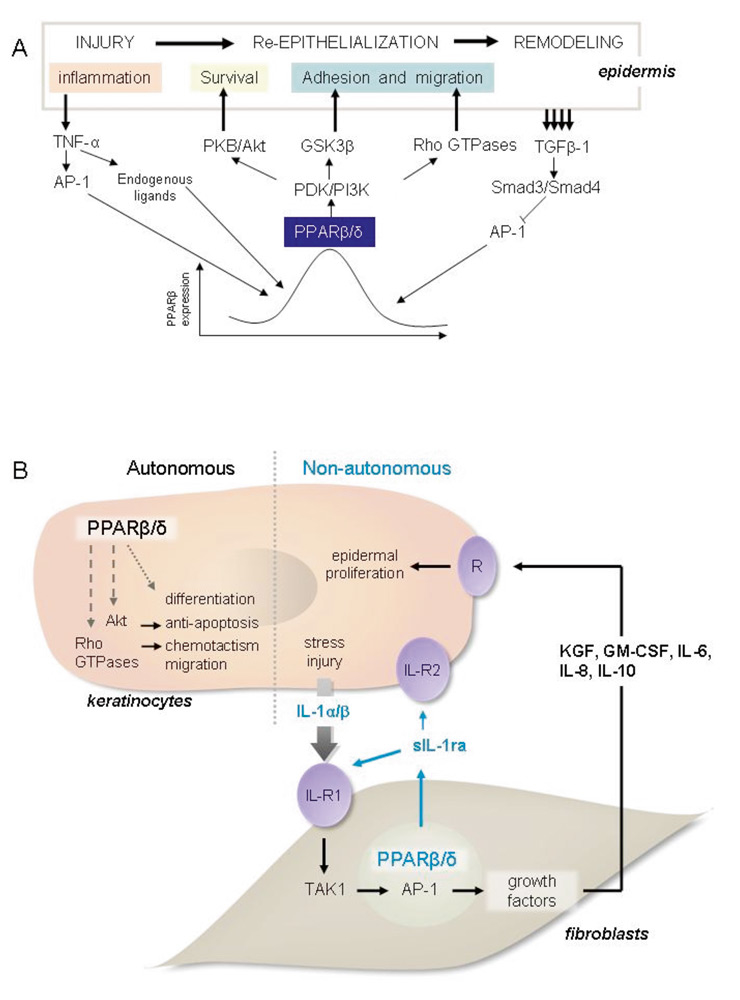
Figure 5
Roles of PPARβ/δ in skin wound healing.
A.) The expression of the pparβ/δ gene is increased via binding of the AP-1 transcription factor complex to its promoter, which is triggered by activation of the stressassociated protein kinase pathway by pro-inflammatory cytokines such as TNF-α. In parallel, the release of pro-inflammatory cytokines also induces the production of PPARβ/δ ligands in the wounded epithelium. The activated PPARβ/δ protein stimulates the genes coding for integrin-linked kinase (ILK) and 3-phosphoinositide-dependent kinase-1 (PDK1), which results in activation of the PKBα/Akt1 anti-apoptotic pathway. Once epithelialisation is completed, TNF-α-induced pparβ/δ expression is repressed by TGFβ-1 signalling which inhibits AP-1 binding to the pparβ/δ promoter.
B.) Cell-autonomous and -nonautonomous effects of PPARβ/δ. PPARβ/δ promote keratinocyte survival differentiation and migration via AKT1 and Rho GTPases in a cell-autonomous manner. Their proliferation is regulated in a non autonomous fashion via a paracrine mechanism. IL-1 that is produced by keratinocytes activates c-Jun in dermal fibroblasts via TAK1. c-Jun is an obligate partner of the AP-1 transcription complex which stimulates the expression of several mitogenic factors. In fibroblasts, PPARβ/δ attenuates IL-1 signalling via the production of sIL-1ra, whose gene is a direct PPARβ/δ target. The sIL-1ra has little affinity for IL-1R2, which is highly expressed in keratinocytes. However, it binds with high affinity to IL-1R1 expressed by the fibroblasts. Therefore, sIL-1ra acts as an autocrine antagonist of IL-1 signalling in fibroblasts. The binding of sIL-1ra to IL-1R1 down-regulates IL-1-mediated signalling events. Consequently, the production of several AP-1-mediated mitogenic factors is down-regulated, which leads to reduced keratinocyte proliferation due to decreased signalling by mitogenic factors mediated by their cognate receptors (R) at the surface of keratinocytes. In brief, PPARβ/δ in fibroblasts has an important homeostatic function in maintaining epidermal homeostasis. The absence of PPARβ/δ results in significant keratinocyte proliferation.
One major result of cellular PPAR action is the balance between cell proliferation, survival and differentiation, as illustrated by the control of key processes such as wound healing. The potential tumourigenic action of PPARs in specific situations deserves important attention, especially when considering the design of a second generation of PPAR agonists.
As lipid sensors, PPARs locally tune gene expression to the metabolic status and thereby coordinate inter-organ communications in developmental, metabolic and immune response processes. The use of mouse models with specific PPAR gain and loss of function in key organs has contributed to a better understanding of the multifaceted roles of PPARs and, in the future, will help unveil their input in the regulatory signalling and gene networks controlling the key functions of these organs. The PPARs still have many secrets to be unveiled, ranging from their molecular actions to their physiological outputs. New technologies allowing genome-wide studies will undoubtedly uncover exciting and intricate actions of PPARs, which hopefully will help in the design of novel targeted pharmacological strategies.
The authors thank Liliane Michalik for comments on the manuscript and Nathalie Constantin for help in preparing the figures.
Correspondence:
Professor
Walter Wahli
Centre for Integrative Genomics
Genopode building
University of Lausanne
Switzerland
Walter.Wahli@unil.ch
References
1 Dandona P, Aljada A, Bandyopadhyay A. Inflammation: the link between insulin resistance, obesity and diabetes. Trends Immunol. 2004;25(1):4–7. Review.
2 Desvergne B, Wahli W. Peroxisome proliferator activated receptors: nuclear control of metabolism. Endocr Rev. 1999;20:649–88.
3 Dreyer C, Krey G, Keller H, Givel F, Helftenbein G, Wahli W. Control of the peroxisomal β-oxidation pathway by a novel family of nuclear hormone receptors. Cell. 1992;68:879–87.
4 Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347:645–50.
5 Nuclear Receptors Nomenclature Committee. A unified nomenclature system for the nuclear receptor superfamily. Cell. 1999;97:1–3.
6 Wahli W. Peroxisome proliferator-activated receptors (PPARs): from metabolic control to epidermal wound healing. Swiss Med Wkly. 2002;132(7-8):83–91.
7 Kota BP, Huang TH, Roufogalis BD. An overview on biological mechanisms of PPARs. Pharmacol Res. 2005;51:85–94.
8 Berger JP, Akiyama TE, Meinke PT. PPARs: therapeutic targets for metabolic disease. Trends Pharmacol Sci. 26(5):244–51.
9 Michalik L, Auwerx J, Berger JP, Chatterjee VK, Glass CK, Gonzalez FJ, et al. International Union of Pharmacology. LXI. Peroxisome proliferator-activator receptors. Pharmacol Rev. 2006;58(4):726–41.
10 Zoete V, Grosdidier A, Michielin O. Peroxisome proliferator-activated receptor structures: ligand specificity, molecular switch and interactions with regulators. Biochim. Biophys Acta. 2007;1771(8):915–25.
11 Yu S, Reddy, JK. Transcription coactivators for peroxisome proliferator-activated receptors. Biochim Biophys Acta. 2007;1771(8):936–51.
12 Guan HP, Ishizuka T, Chui PC, Lehrke M, Lazar MA. Corepressors selectively control the transcriptional activity of PPARγ in adipocytes. Genes Dev. 2005;19(4):453–61.
13 Leuenberger N, Pradervand S, Wahli W. Sumoylated PPARalpha mediates sex-specific gene repression and protects the liver from estrogen-induced toxicity in mice. J Clin Invest. 2009;119(10):3138–48.
14 Michalik L, Wahli W. Involvement of PPAR nuclear receptors in tissue injury and wound repair. J Clin Invest. 2006;116(3):598–606.
15 Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, et al. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317(5834):121–4.
16 Bosch FX., Ribes J, Diaz M, Cleries R. Primary liver cancer: worldwide incidence and trends. Gastroenterology. 2004;127:S5-16.
17 Crockett ET, Spielman W, Dowlatshahi S, He J. Sex differences in inflammatory cytokine production in hepatic ischemia-reperfusion. J Inflamm. (Lond.). 2006;3:16.
18 Mittendorfer B. Sexual dimorphism in human lipid metabolism. J Nutr. 2005;135:681–6.
19 Anzenbacher P, Anzenbacherova E. Cytochromes P450 and metabolism of xenobiotics. Cell Mol Life Sci. 2001;58:737–47.
20 Wang H, Zhao Y, Bradbury JA, Graves JP, Foley J, Blaisdell JA, et al. Cloning, expression, and characterization of three new mouse cytochrome p450 enzymes and partial characterization of their fatty acid oxidation activities. Mol Pharmacol. 2004;65(5):1148–58.
21 Lamba V, Lamba J, Yasuda K, Strom S, Davila J, Hancock ML, et al. Hepatic CYP2B6 expression: gender and ethnic differences and relationship to CYP2B6 genotype and CAR (constitutive androstane receptor) expression. J Pharmacol Exp Ther. 2003;307(3):906–22.
22 Stapleton G, Steel M, Richardson M, Mason JO, Rose KA, Morris RG, et al. A novel cytochrome P450 expressed primarily in brain. J Biol Chem. 1995;270(50):29739–45.
23 Martin C, Bean R, Rose K, Habib F, Seckl J. Cyp7b1 catalyses the 7alpha-hydroxylation of dehydroepiandrosterone and 25-hydroxycholesterol in rat prostate. Biochem J. 2001;355:509–15.
24 Pettersson H, Holmberg L, Axelson M, Norlin M. CYP7B1-mediated metabolism of dehydroepiandrosterone and 5-alpha-androstane -3beta,17beta-diol--potential role(s) for estrogen signaling. FEBS J. 2008;275:1778–89.
25 Tang W, Eggertsen G, Chiang JY, Norlin M. Estrogen-mediated regulation of CYP7B1: a possible role for controlling DHEA levels in human tissues. J Steroid Biochem Mol Biol. 2006;100:42–51.
26 Umetani M, Domoto H, Gormley AK, Yuhanna IS, Cummins CL, Javitt NB, et al. 27-Hydroxycholesterol is an endogenous SERM that inhibits the cardiovascular effects of estrogen. Nat Med. 2007;13(10):1185–92.
27 Yamamoto Y, Moore R, Hess HA, Guo GL, Gonzalez FJ, Korach KS, et al. Estrogen receptor alpha mediates 17alpha-ethynylestradiol causing hepatotoxicity. J Biol Chem. 2006;281:16625–31.
28 Pusl T, Beuers U. Intrahepatic cholestasis of pregnancy. Orphanet J Rare Dis. 2007;2:26.
29 Michalik L, Desvergne B, Wahli W. Peroxisome proliferator-activated receptors and cancers: complex stories. Nature Rev Cancer. 2004;4(1):61–70.
30 Rotman N, Michalik L, Desvergne B, Wahli W. PPARs in fetal and early postnatal development. Adv Develop Biol. 2006;16:33–64.
31 Hashimoto T, Cook WS, Qi C, Yeldandi AV, Reddy JK, Rao MS. Defect in peroxisome proliferator-activated receptor alpha-inducible fatty acid oxidation determines the severity of hepatic steatosis in response to fasting. J Biol Chem. 2000;275:28918–28.
32 Kersten S, Seydoux J, Peters JM, Gonzalez FJ, Desvergne B, Wahli W. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J Clin Invest. 1999;103:1489–98.
33 Leone TC, Weinheimer CJ, Kelly DP. A critical role for the peroxisome proliferator-activated receptor alpha (PPARalpha) in the cellular fasting response: the PPARalpha-null mouse as a model of fatty acid oxidation disorders. Proc Natl Acad Sci. USA 1999;96:7473–8.
34 Martin G, Schoonjans K, Lefebvre AM, Staels B, Auwerx J. Coordinate regulation of the expression of the fatty acid transport protein and acyl-CoA synthetase genes by PPARalpha and PPARgamma activators. J Biol Chem. 1997;272:28210–7.
35 Sato O, Kuriki C, Fukui Y, Motojima K. Dual promoter structure of mouse and human fatty acid translocase/CD36 genes and unique transcriptional activation by peroxisome proliferator-activated receptor alpha and gamma ligands. J Biol Chem. 2002;277:15703–11.
36 Crisafulli C, Bruscoli S, Esposito E, Mazzon E, Di Paola R, Genovese T, et al. PPAR-alpha contributes to the anti-inflammatory activity of 17beta-estradiol. J Pharmacol Exp Ther. 2009;331(3):796–807.
37 Lemberger T, Saladin R, Vázquez M, Assimacopoulos F, Staels B, Desvergne B, et al. Expression of the peroxisome proliferator-activated receptor alpha gene is stimulated by stress and follows a diurnal rhythm. J Biol Chem. 1996;271(3):1764–9.
38 Rotman N, Wahli W. Fatty acid synthesis and PPARalpha hand in hand. Chem Biol. 2009;16(8):801–2.
39 Chakravarthy MV, Lodhi IJ, Yin L, Malapaka RR, Xu HE, Turk J, et al. Identification of a physiologically relevant endogenous ligand for PPARalpha in liver. Cell. 2009;138(3):476–88.
40 Yessoufou A, Hichami A, Besnard P, Moutairou K, Khan NA. Peroxisome proliferator-activated receptor alpha deficiency increases the risk of maternal abortion and neonatal mortality in murine pregnancy with or without diabetes mellitus: Modulation of T cell differentiation. Endocrinology. 2006;147(9):4410–8.
41 Hashimoto F, Oguchi Y, Morita M, Matsuoka K, Takeda S, Kimura M, et al. PPAR-alpha agonists clofibrate and gemfibrozil inhibit cell growth, down-regulate hCG and up-regulate progesterone secretions in immortalized human trophoblast cells. Biochem Pharmacol. 2004;68:313–21.
42 Michalik L, Desvergne B, Dreyer C, Gavillet M, Laurini RN, Wahli W. PPAR expression and function during vertebrate development. Int J Dev Biol. 2002;46:105–14.
43 Wang Q, Fujii H, Knipp GT. Expression of PPAR and RXR isoforms in the developing rat and human term placentas. Placenta. 2002;23:661–71.
44 Tontonoz P, Hu E, Graves R, Budavari A, Spiegelman B. mPPAR gamma 2: tissue-specific regulator of an adipocyte enhancer. Genes Dev. 1994;8(10):1224–34.
45 Forman B, Tontonoz P, Chen J, Brun R, Spiegelman B, Evans R. 15-Deoxy-delta 12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPAR gamma. Cell. 1995;83:803–12.
46 Patsouris D, Reddy JK, Muller M, Kersten S. Peroxisome proliferator-activated receptor alpha mediates the effects of high-fat diet on hepatic gene expression. Endocrinology. 2006;147:1508–16.
47 Yu S, Matsusue K, Kashireddy P, Cao WQ, Yeldandi V, Yeldandi AV, et al. Adipocyte-specific gene expression and adipogenic steatosis in the mouse liver due to peroxisome proliferator-activated receptor gamma1 (PPARgamma1) overexpression. J Biol Chem. 2003;278:498–505.
48 Wahli W. PPAR gamma: ally and foe in bone metabolism. Cell Metab. 2008;7(3):188–90.
49 Akune T, Ohba S, Kamekura S, Yamaguchi M, Chung UI, Kubota N, et al. PPARgamma insufficiency enhances osteogenesis through osteoblast formation from bone marrow progenitors. J Clin Invest. 2004;113:846–55.
50 Cock TA, Back J, Elefteriou F, Karsenty G, Kastner P, Chan S, et al. Enhanced bone formation in lipodystrophic PPARgamma(hyp/hyp) mice relocates haematopoiesis to the spleen. EMBO Rep. 2004;5:1007–12.
51 Nadra K, Anghel SI, Joye E, Tan NS, Basu-Modak S, Trono D, et al. Differentiation of trophoblast giant cells and their metabolic functions are dependent on peroxisome proliferator-activated receptor β/δ. Mol Cell Biol. 2006;26:3266–81.
52 Barak Y, Nelson MC, Ong ES, Jones YZ, Ruiz-Lozano P, Chien KR, et al. PPAR gamma is required for placental, cardiac, and adipose tissue development. Mol Cell. 1999;4:585–95.
53 Barish GD, Narkar VA, Evans RM. PPAR delta: a dagger in the heart of the metabolic syndrome. J Clin Invest. 2006;116:590–7.
54 Lim H, Dey SK. PPAR delta functions as a prostacyclin receptor in blastocyst implantation. Trends Endocrinol Metab. 2000;11:137–42.
55 Peters JM, Lee SS, Li W, Ward JM, Gavrilova O, Everett C, et al. Growth, adipose, brain, and skin alterations resulting from targeted disruption of the mouse peroxisome proliferator-activated receptor beta (delta). Mol Cell Biol. 2000;20:5119–28.
56 Mano H, Kimura C, Fujisawa Y, Kameda T, Watanabe-Mano M, Kaneko H, et al. Cloning and function of rabbit peroxisome proliferator-activated receptor delta/beta in mature osteoclasts. J Biol Chem. 2000;275:8126–32.
57 Hansen JB, Zhang H, Rasmussen TH, Petersen RK, Flindt EN, Kristiansen K. Peroxisome proliferator-activated receptor-δ (PPARδ)-mediated regulation of preadipocyte proliferation and gene expression is dependent on cAMP signaling. J Biol Chem. 2001;276:3175–82.
58 Schuler M, Ali F, Chambon C, Duteil D, Bornert JM, Tardivel A, et al. PGC1alpha expression is controlled in skeletal muscles by PPARbeta, whose ablation results in fiber-type switching, obesity, and type 2 diabetes. Cell Metab. 2006;4(5):407–14.
59 Oliver WR Jr, Shenk JL, Snaith MR, Russell CS, Plunket KD, Bodkin NL, et al. A selective peroxisome proliferator-activated receptor delta agonist promotes reverse cholesterol transport. Proc Natl Acad Sci. USA 2001;98:5306–11.
60 Krey G, Braissant O, L’Horset F, Kalkhoven E, Perroud M, Parker MG, et al. Fatty acids, eicosanoids, and hypolipidaemic agents identified as ligands of peroxisome proliferator-activated receptors by coactivator-dependent receptor ligand assay. Mol Endocrinol. 1997;11:779–91.
61 Escher P, Braissant O, Basu-Modak S, Michalik L, Wahli W, Desvergne B. Rat PPARs: quantitative analysis in adult rat tissues and regulation in fasting and refeeding. Endocrinology. 2001;142:4195–202.
62 Hoekstra M, Kruijt JK, Van Eck M, Van Berkel TJ. Specific gene expression of ATP-binding cassette transporters and nuclear hormone receptors in rat liver parenchymal, endothelial, and Kupffer cells. J Biol Chem. 2003;278:25448–53.
63 Shan W, Nicol CJ, Ito S, Bility MT, Kennett MJ, Ward JM, et al. Peroxisome proliferator-activated receptor-beta/delta protects against chemically induced liver toxicity in mice. Hepatology. 2008;47:225–35.
64 Sanderson LM, Boekschoten MV, Desvergne B, Müller M, Kersten S. Transcriptional profiling reveals divergent roles of PPARalpha and PPARbeta/delta in regulation of gene expression in mouse liver. Physiol Genomics. 2010;41(1):42–52.
65 Ravnskjaer K, Frigerio F, Boergesen M, Nielsen T, Maechler P, Mandrup S. PPARδ is a fatty acid sensor, which enhances mitochondrial oxidation in insulin-secreting cells and protects against fatty acid induced dysfunction. J Lipid Res. 2009 Nov 30. [Epub ahead of print] PMID: 19965574.
66 Coll T, Rodrïguez-Calvo R, Barroso E, Serrano L, Eyre E, Palomer X, et al. Peroxisome proliferator-activated receptor (PPAR) beta/delta: a new potential therapeutic target for the treatment of metabolic syndrome. Curr Mol Pharmacol. 2009;2(1):46–55.
67 Kielian T, Drew PD. Effects of perixosome proliferator activated receptor-g agonists on central nervous system inflammation. J Neurosci Res. 2003;71:315–25.
68 Lovett-Racke AE, Hussain RZ, Northrop S, Choy J, Rocchini A, Matthes L, et al. Peroxisome proliferator-activated receptor alpha agonists as therapy for autoimmune disease. J Immunol. 2004;172:5790–8.
69 Dubuquoy L, Rousseaux C, Thuru X, Peyrin-Biroulet L, Romano O, Chavatte P, et al. PPARgamma as a new therapeutic target in inflammatory bowel diseases. Gut. 2006;55:1341–9.
70 Polak PE, Kalinin S, Dello Russo C, Gavrilyuk V, Sharp A, Peters JM, et al. Protective effects of a peroxisome proliferator activated receptor-b/d agonist in experimental autoimmune encephalomyelitis. J Neuroimmunol. 2005;168:65–75.
71 Attakpa E, Hichami A, Simonin AM, Sansón EG, Dramane KL, Khan NA. Docosahexaenoic acid modulates the expression of T-bet and GATA-3 transcription factors, independently of PPARalpha, through suppression of MAP kinase activation. Biochimie. 2009;91(11-12):1359–65.
72 Delerive P, De Bosscher K, Besnard S, Vanden Berghe W, Peters JM, Gonzalez FJ, et al. Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-κB and AP-1. J Biol Chem. 1999;274:32048–54.
73 Takano H, Hasegawa H, Nagai T, Komuro I. The role of PPARγ dependent pathway in the development of cardiac hypertrophy. Drugs Today. (Barc) 2003;39:347–57.
74 Shah YM, Morimura K, Gonzalez FJ. Expression of peroxisome proliferator activated receptor-g in macrophage suppresses experimentally induced colitis. Am J Physiol Gastrointest Liver Physiol. 2007;292:G657–66.
75 Adachi M, Kurotani R, Morimura K, Shah Y, Sanford M, Madison BB, et al. Peroxisome proliferator activated receptor gamma in colonic epithelial cells protects against experimental inflammatory bowel disease. Gut. 2006;55:1104–11.
76 Straus DS, Glass CK. Anti-inflammatory actions of PPAR ligands: new insights on cellular and molecular mechanisms. Trends Immunol. 2007;28:551–8.
77 Okamoto H, Iwamoto T, Kotake S, Momohara S, Yamanaka H, Kamatani N. Inhibition of NF-κB signaling by fenofibrate, a peroxisome proliferator-activated receptor-α ligand, presents a therapeutic strategy for rheumatoid arthritis. Clin Exp Rheumatol. 2005;23:323-30.
78 Madej A, Okopien B, Kowalski J, Zielinski M, Wysocki J, Szygula B, et al. Effects of fenofibrate on plasma cytokine concentrations in patients with atherosclerosis and hyper-lipoproteinemia IIb. Int J Clin Pharmacol Ther. 1998;36:345–9.
79 Michalik L, Wahli W. PPARs mediate lipid signaling in inflammation and cancer. PPAR Res. 2008;2008:1–15.
80 Michalik L, B. Desvergne B, Tan NS, Basu-Modak S, Escher P, Rieusset J, et al. Impaired skin wound healing in peroxisome proliferator-activated receptor (PPAR)α and PPARβ mutant mice. J Cell Biol. 2001;154:799–814.
81 Tan NS, Michalik L, Noy N, Yasmin R, Pacot C, Heim M, et al. Critical roles of PPAR beta/delta in keratinocyte response to inflammation. Genes Dev. 2001;15:3263–77.
82 Michalik L, Feige JN, Gelman L, Pedrazzini T, Keller H, Desvergne B, et al. Selective expression of a dominant-negative form of peroxisome proliferator-activated receptor in keratinocytes leads to impaired epidermal healing. Mol Endocrinol. 2005;19:2335–48.
83 Chong HC, Tan MJ, Philippe V, Tan SH, Tan CK, Ku CW, et al. Regulation of epithelial-mesenchymal IL-1 signaling by PPARbeta/delta is essential for skin homeostasis and wound healing. J Cell Biol. 2009;184(6):817–31.
84 Hanley K, Jiang Y, Crumrine D, Bass NM, Appel R, Elias PM, et al. Activators of the nuclear hormone receptors PPARalpha and FXR accelerate the development of the fetal epidermal permeability barrier, J Clin Invest. 1997;100:705–12.
85 Mao-Qiang M, Fowler AJ, Schmuth M, Lau P, Chang S, Brown BE, et al. Peroxisome-proliferator-activated receptor (PPAR)-gamma activation stimulates keratinocyte differentiation. J Invest Dermatol. 2004;123:305–12.