Liver injury caused by drugs: an update
DOI: https://doi.org/10.4414/smw.2010.13080
G
Stirnimann, K
Kessebohm
Summary
Although severe idiosyncratic drug–induced liver injury (DILI) is a rare event, it has a large impact on the fate of affected patients and the incriminated drug. Hepatic metabolism of drugs, which occurs in the generation of chemically reactive metabolites in critical amounts, seems to underlie most instances of DILI. Genetic polymorphisms in activating and detoxifying enzymes determine, in part, the extent of cellular stress. A cascade of events, where the pathogenetic relevance of single steps is likely to vary from drug to drug, leads to the disturbance of cellular homeostasis, to mitochondrial dysfunction, to the activation of cell death promoting pathways and the release of drug-modified macromolecules and/or danger signals that initiate an innate and/or adaptive immune response. The patient’s response to the initial drug-induced cellular dysfunction determines whether adaptation to the drug-induced cellular stress or DILI in one of its many forms of clinical presentation occurs. Although risk factors for developing DILI have been identified and many pathogenetic mechanisms have been elucidated in model systems, idiosyncratic drug reactions remain unpredictable.
Introduction
Our predictors may be good at predicting the ordinary, but not the irregular, and this is where they ultimately fail (N. N. Taleb, The Black Swan).
Severe drug–induced liver injury (DILI) leading to liver failure, transplantation or death is a rare event, and is – with few exceptions – unpredictable and poorly understood regarding its pathogenesis. Nevertheless, it represents an important health problem: Although the incidence of idiosyncratic DILI with approved drugs in therapeutic doses is relatively low and estimated at 1 per 10 000 to 1 per 100 000 treated patients, every seventh case of acute hepatic failure is due to an adverse drug reaction and DILI has become the leading cause for super-urgent liver transplantation [1–3]. It is also the most common adverse event that halts the development of a new drug or leads to the withdrawal of approved drugs from the market. Due to the low incidence of DILI, a drug’s hepatotoxic potential is usually not recognised during pre-marketing trials and only manifests itself after the drug has been marketed and used by many more patients than are included in clinical trials [4, 5]. Over 1000 drugs and herbal products have been associated with idiosyncratic hepatotoxicity [6, 7].
Idiosyncrasy refers to inter-individual differences in the response to stimuli due to genetic and environmental factors. Not all treated patients respond to a particular drug and only a fraction develops severe adverse effects. ‘Idiosyncratic’ does not imply dose independentnor does it necessarily mean rare, but rather it indicates a reaction that may not be seen regularly. Nevertheless, it is a rare event in clinical practice because drugs causing idiosyncratic DILI with an incidence high enough to be detected during clinical development usually do not make it to the market. Whereas traditional pharmacological and toxicological concepts focus on dosing and effects in the average patient, one has to concentrate on unusual patterns of drug handling and response, i.e., the statistical outliers, if one intends to better understand, prevent and treat idiosyncratic drug reactions.
Clinical presentation and diagnosis
The clinical presentation of DILI may vary substantially. It can mimic other known forms of acute and chronic liver diseases and the severity may range from asymptomatic elevations of hepatic enzymes to fulminant hepatic failure. Somewhat arbitrary, the liver injury is classified as hepatocellular, mixed or cholestatic depending on the ratio of alanine aminotransferase to alkaline phosphatase [8]. These biomarkers indicate liver injury but are not specific for drug-induced injury which often remains a diagnosis based on the exclusion of other known causes of acute or chronic liver diseases. Histology is generally not helpful in regard to possible drug aetiology but may help exclude other suspected diagnoses. To diagnose DILI with any degree of certainty can be difficult and must take into account factors such as the temporal relationship between drug exposure and clinical events, more or less characteristic patterns of clinical presentation of injury related to specific drugs, and effects of de-challenge and – often inadvertent – re-challenge. Considering that many drugs can present with different patterns of injury in different individuals further complicates the diagnostic process. For example, in a recent report of 102 cases with amoxicillin/clavulanic acid associated liver injury that is generally thought to present as cholostatic hepatitis, a third presented with acute hepatocelluar injury, another third were mixed and the remaining third with cholestatic injury [9]. Algorithms have been developed to help in causality assessments, for example the Roussel Uclaf Causality Assessment Model [10], but they are by no means perfect and are rather complicated to use in practice, so that expert opinion remains the gold standard [11]. An improved causality assessment method based on a prospective registry is being developed by a group of experts participating in the multicentre Drug-Induced Liver Injury Network [12].
Specific diagnostic tests for DILI have been proposed for selected drugs but are not generally applicable, nor are they generally available. The detection of circulating drug-protein adducts can be of assistance in the diagnosis of paracetamol hepatotoxicity, but may also be present in healthy subjects taking paracetamol [13]. Antibodies against drug-protein adducts or drug-metabolising enzymes can be found in patients with liver injury due to some drugs, such as diclofenac, tienilic acid, dihydralazine and halothane, but their diagnostic value is not established. A lymphocyte transformation test, where the patient’s lymphocytes are cultured in the presence of the drug suspected to have caused the adverse reaction, may help establish causality in some cases of DILI. Whether metabolomics, proteomics, genomics or transcriptomics will eventually be of assistance in the diagnosis of DILI remains to be demonstrated.
About 10% of the patients with severe DILI associated with jaundice will either die or need liver transplantation [14]. When a patient has survived an episode of severe DILI, clinically relevant liver disease rarely ensues. However, especially cholestatic/mixed type of liver injury can take a protracted course [15]. Approximately 1%, mainly patients who initially presented with hepatocellular type of DILI, will go on to develop cryptogenic cirrhosis. The risk of evolving to chronic liver disease appears to be higher in patients who were treated for a longer time with the incriminated drug. Continuing treatment with a drug that has begun to cause liver injury may increase the risk of not only developing more severe acute damage but also chronic injury [16, 17]. About a fifth of the patients who present with chronic liver disease after an episode of DILI have a bona fide auto-immune hepatitis [15]. Causality is difficult to establish, particularly since cryptogenic cirrhosis and auto-immune hepatitis are not rare entities. It may be that liver injury due to a drug increases the susceptibility to subsequently develop auto-immune hepatitis and cirrhosis, respectively.
Risk factors
An idiosyncratic drug reaction does not imply that it occurs independent of dose. At first glance, it may appear so because these drug reactions usually occur with recommended therapeutic and not with excessively high doses. Nevertheless, the risk of DILI is, in part, determined by the amount of the administered drug (i.e., the daily dose). Data from Sweden and the United States show that regardless of the drug taken, three quarters of cases of DILI follow exposure to drugs with recommended daily doses of 50 mg or more and only 9% of the cases occur with drugs at daily doses of less than 10 mg [14, 18]. Of 111 liver transplantations performed for hepatic failure due to an oral prescription drug other than paracetamol, 101 were done in patients who took a drug at a dose of >50 mg, 8 in patients with drugs at doses of 11–49 mg, and only 2 in patients with drugs that were taken at a daily dose of <10 mg [19]. Not only the daily dose but also the extent of hepatic metabolism is an important determinant of the risk of liver injury. Of the 200 most frequently prescribed drugs, the ones with >50% hepatic metabolism resulted more frequently in elevations of transaminases to more than three times the upper limit of normal and liver failure than drugs with little or no hepatic metabolism [20]. These observations support the hypothesis that most cases of severe DILI are due to the formation of reactive metabolites and that only limited and thus generally non-toxic amounts of reactive metabolites can be formed from a drug that is taken at daily doses of 10 mg or less [21].
Whether increasing age and female gender confers an increased risk for the development of DILI is controversial. Recent, prospectively collected data from Spain indicates that the incidence of DILI is not higher in females and does not increase with increasing age. However, cholestatic injury may occur more frequently in men over 60 and hepatocellular injury may occur more frequently in younger women [9]. Several reports have indicated an increased risk for isoniazid-associated liver injury in patients above the age of 60 (for review see Tostmann et al. [22]). Age was not a risk factor in other case series and in a recent prospective study, including 70 patients with acute liver failure associated with antituberculous therapy, the median age was 27 with 70% of the patients under the age of 35 [23]. The risk, if any, conferred by age does certainly not preclude an indicated drug therapy. Patients with chronic liver diseases are generally not at a higher risk of developing drug-induced
injury, with the possible exception of patients with viral hepatitis who may be more susceptible to anti-tuberculous and anti-retroviral drugs [24, 25]. Although patients with chronic liver disease may have impaired defence mechanisms, such as low intrahepatic concentrations of glutathione or low activity of superoxide dismutase, they may on the other hand be protected because the activity of enzymes generating potentially toxic metabolites is decreased. Patients with cirrhosis and a compromised functional reserve may, however, have a poorer outcome should they develop DILI because their livers do not tolerate the further loss in function associated with an episode of DILI.
Mechanistic hypotheses
Mechanistic hypotheses are difficult to test without reproducible animal models. Unfortunately, animal models for idiosyncratic liver injury are scarce because these reactions are idiosyncratic in animals as well. Some investigators have used animals which are made more susceptible to drug-induced injury by either introducing an underlying inflammatory response or by removing protective genes, resulting for example in a decreased activity of superoxide dismutase [26–28]. The clinical relevance of these models is not established and it is not known whether they may reliably predict the risk of a newly developed drug for idiosyncratic liver injury or help establish rational therapeutic interventions once drug injury has occurred.
This is different for the best, by far, studied model of paracetamol hepatotoxicity, which is clearly, however, not an idiosyncratic reaction. Based on this model, a good understanding exists of how a cascade of events initiated by a reactive metabolite of a drug can result in the death of a hepatocyte. Some of the events that are relevant in paracetamol toxicity have also been demonstrated in in vitrosystems with drugs causing idiosyncratic hepatotoxicity, suggesting that lessons learned from paracetamol may help our understanding of idiosyncratic drug reactions.
Paracetamol hepatotoxicity
Paracetamol is a common cause of acute hepatic failure but differs from most other drugs resulting in DILI in that its hepatotoxicity is dose related and reproducible in animals. In the early 1970s, Jerry Mitchell and collaborators showed that the toxicity of paracetamol is due to metabolic activation of the drug to a toxic metabolite that is preferentially conjugated with glutathione, a process many-fold accelerated by the activity of glutathione transferase [29]. Once glutathione is depleted, the toxic metabolite, N-acetyl-p-benzoquinone imine, binds to other nucleophilic groups in the cell. Cell death ensues, especially in zone III hepatocytes around the central vein, but also in non-liver cells able to activate the parent compound, such as renal tubular cells. The central role of activation by cytochrome-P450 (CYP450) and detoxification by glutathione is clearly demonstrated by the protection against hepatotoxicity which can be attained by inhibition of CYP450 activity and by replenishing glutathione stores. Indeed, paracetamol toxicity is a prime example of a clinical problem that was elucidated in animal models and, once solved, successfully changed clinical practice with a rational therapeutic intervention. Today, supplementation of precursor amino acids for glutathione synthesis, most commonly N-acetyl-cysteine, is standard therapy for all patients suspected of a paracetamol overdose [30]. The critical role of glutathione may explain why patients with decreased glutathione stores prior to paracetamol exposure may be at increased risk of developing hepatic injury. Two such groups are
alcoholics and malnourished patients who have lower reserves of glutathione [31]. Patients consuming excessive amounts of ethanol and consequently induced CYP2E1, the cytochrome mainly responsible for the activation of paracetamol, may develop severe toxicity from paracetamol following doses that are considered safe [32].
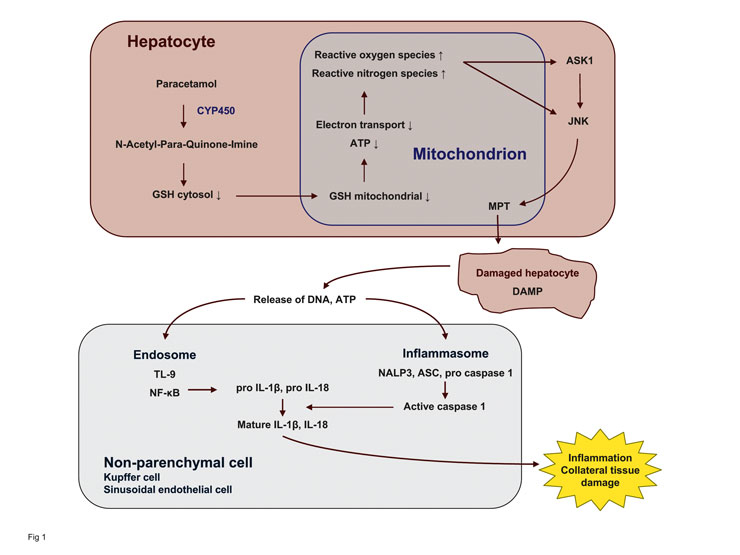
Figure 1
Model of paracetamol hepatotoxicity. Paracetamol impairs mitochondrial function via formation of a reactive metabolite and depletion of glutathione. The resulting oxidant stress activates a redox-sensitive kinase (ASK1) and subsequently a cell-death promoting kinase (JNK). The latter translocates to mitochondria and promotes the mitochondrial permeability transition (MPT). The damaged hepatocytes release danger signals (danger associated molecular pattern, DAMP) which via their interaction with the endosome and inflammasome generate an innate immune response by non-parenchymal cells.
Depletion of glutathione and covalent binding to liver proteins per se is not sufficient to lead to cell death. A regioisomer of paracetamol, 3’-hydroxyacetanilide (AMAP), depletes hepatic glutathione and binds to proteins to the same extent as paracetamol but does not result in toxicity. One key difference between the two compounds is that AMAP spares mitochondria whereas paracetamol also depletes mitochondrial glutathione [33]. Mitochondrial glutathione has to be transported from the cytosol into mitochondria which themselves do not synthesise the tripeptide [34]. Interestingly, alcohol preferentially depletes mitochondrial glutathione which may be relevant for the increased susceptibility of alcoholics to low doses of paracetamol. Depletion of mitochondrial glutathione and the resulting oxidant stress is followed by redox-sensitive
activation of c-jun-N-terminal kinases (JNK) [35] leading to the opening of the mitochondrial permeability transition pore (fig. 1). Inhibition of JNK or the silencing of JNK in vivo protects against the toxicity of paracetamol without interfering with its metabolism and the depletion of glutathione, indicating that metabolic activation and depletion of glutathione is a prerequisite to trigger a host response leading to cell death but is not lethal itself [36, 37].
Liver injury by paracetamol is accompanied by an innate immune response as shown by the
up-regulation of the inflammatory cytokine interleukin-1β [38]. Free DNA released from dying hepatocytes engages toll-like receptor 9 (TLR9) in liver sinusoidal endothelial cells and possibly Kupffer cells, leading to the NF-κB-dependent production of cytokine precursors [39]. Concurrent activation of the NALP3 inflammasome (a
multiprotein complex responsible for activating caspases), possibly triggered by ATP or uric acid
released from necrotic hepatocytes, leads to the
activation of caspase-1 which proteolytically cleaves pro-IL-1β and pro-IL-18 into their active forms. Knock-out mice lacking elements of the inflammasome pathway (caspase-1, NALP3) and animals treated with inhibitors of TLR9 or interleukins are protected against paracetamol injury in vivo.
Taken together, the available data indicate that paracetamol by its metabolism to a reactive metabolite results in covalent binding and depletion of glutathione. This sensitises mitochondria to JNK-induced mitochondrial permeability transition that leads to cell death, which in turn promotes immune activation by generating a danger-associated molecular pattern (DAMP) that stimulates cytokine production in neighbouring sinusoidal endothelial cells via TLR9 and the NALP3 inflammasome. The relevance of the sequence of events is supported by successful therapeutic interventions at different steps in vivo. Paracetamol hepatotoxicity can be prevented in intact animals (and also in humans in part) by inhibiting the activation of paracetamol to a reactive metabolite by CYP450, by stimulating the formation of detoxifying glutathione, by interfering with the JNK pathway and finally by preventing immune activation (i.e., the host response to hepatocyte injury). Whether the described mechanisms are unique to paracetamol or whether they provide a model for other idiosyncratic drug reactions remains to be demonstrated. Some elements of the cascade of events resulting in paracetamol-induced hepatic failure have also been demonstrated with drugs involved in DILI in model systems, suggesting that similar mechanisms may, in part, be involved in idiosyncratic DILI.
Central role of mitochondria
Mitochondrial dysfunction, which is a key element in paracetamol toxicity, may also play a role in liver injury from other drugs that increase the formation of reactive oxygen species in mitochondria and may sensitise mitochondria to stress kinases [40, 41]. The presence of microvesicular steatosis in liver biopsies of some patients with suspected DILI suggests that mitochondrial dysfunction is an early event in these cases. Many drugs that are associated with idiosyncratic DILI in humans affect mitochondrial function, deplete cellular ATP or induce mitochondrial permeabilisation in model systems, including diclofenac, troglitazone, nimesulide, mefenamic acid, amiodarone, benzbromarone, trovafloxacin, lamivudine, valproic acid and tolcapone (for references see [41]).
Assuming that injury to mitochondria is pivotal in precipitating DILI is an attractive hypothesis. As exemplified by paracetamol, injury to mitochondria can mediate cell death by the release of death proteins, such as cytochrome c, endonuclease G, and Smac/Diablo, which in turn will translocate to the nucleus or activate a cascade of proteases leading to apoptosis or necrosis. Drug-induced oxidative stress can lead to the activation of cell-death activating pathways without depletion of glutathione. Furthermore, progressive mitochondrial damage that does not manifest itself until a threshold is reached where the death program is set in motion could explain the delayed onset typically seen in DILI. The mitochondrial DNA (mtDNA) which encodes for several subunits of the electron transfer complexes is more susceptible to oxidative damage than nuclear DNA because mtDNA is close to the site of generation of reactive oxygen species, it lacks protective histones, the repair system is not efficient and there are few non-coding sequences. Thus, a cell may accumulate an increasing number of mitochondria with an increasing extent of DNA damage until the cumulative damage triggers the death of the cell. Progressive mitochondrial dysfunction resulting in hepatic failure occurs with nucleoside analogues used in the treatment of viral infections that are incorporated into mtDNA instead of physiologic nucleosides [42]. Since humans accumulate damaged mtDNA as they age, this might provide an explanation for an increased susceptibility of older patients to some drugs causing liver
injury. In addition to acquired damage to mtDNA, inherited mutations therein could increase the
susceptibility to drug injury. There are case reports suggesting that pre-existing mitochondrial impairment and certain polymorphisms in the mitochondrial DNA polymerase gamma gene increase the risk to valproic acid associated liver failure [43].
Animal models for DILI have been developed based on an impaired defence against mitochondrial oxidative stress. Troglitazone is not toxic in normal animals. However, in mice deficient in superoxide dismutase (SOD2
+/–
) whose mitochondrial antioxidant defence is impaired, it results in an increase in serum transaminases after four weeks of exposure [27]. This time course mimics the clinical course of hepatotoxicity of troglitazone which has been withdrawn from the market because of this adverse effect.
Adaptation
The phenomenon of adaptation was first observed in the 1970s in large trials with isoniazid (INH). It was noted that 20–30% of the people treated with INH for prophylaxis of tuberculosis showed moderate elevations of transaminases that subsequently normalised in spite of continuing the drug [44]. Only a small portion of the patients went on to develop severe liver injury. Thus, most people are seemingly able to adapt to the damaging effects of the drug. Transient elevations of transaminases are also seen with troglitazone [45], tacrine [46] and ximelagatran [47]. The mechanisms of adaptation are poorly understood. They may involve up- and down-regulation of transporters [48, 49] and drug metabolising enzymes [50], induction of mitochondrial biogenesis, development of immune tolerance [51] and finally regeneration.
Genetic associations
Genetic differences in the activation of a drug to a toxic intermediate and its detoxification could explain inter-individual differences in susceptibility and thus the idiosyncratic nature of DILI. The search for relevant genetic factors, however, is hampered by the low number of index cases. So far, most investigations have focussed on known genes that are involved in drug disposition, in oxidative stress, or in the immune response. Only recently has an unbiased approach been possible by performing genome-wide association studies [52].
Isoniazid leads to a moderate increase in transaminases in about 20% of the patients, most of whom are able to adapt and normalise their liver enzyme despite continuation of the drug. About 3% of the patients, however, progress to more severe liver injury. Metabolites of INH, acetylhydrazine and hydrazine, are thought to be responsible for the liver injury that can be reproduced to some extent in experimental animals. The exposure to these two metabolites following the administration of INH depends on the activity of N-acetyltransferase (NAT-2) [53, 54] which is one of the first drug-metabolising enzymes for which a polymorphism was recognised. Several studies have shown that the risk of developing liver injury from INH is increased in slow acetylators [55], particularly if INH is taken together with rifampicin which is a potent inducer of cytochrome P-450 enzymes and might thus increase the formation of toxic metabolites [56, 57]. Alcoholic patients appear to be more susceptible to INH hepatotoxicity. Ethanol induces the cytochrome P450 isoenzyme, CYP2E1, which is also responsible for the activation of acetylhydrazine to toxic metabolites. Taiwanese patients with wild type CYP2E1 had a higher risk of INH hepatotoxicity than patients carrying the CYP2E1*5 allele [58]; a finding that could not, however, be confirmed in Korean patients [59].
Diclofenac, like other non-steroidal anti-inflammatory drugs, can result in DILI and has been studied extensively in regard to possible mechanisms. Diclofenac is metabolised by CYP2C9 and also undergoes glucuronidation by uridine diphosphate glucuronosyltransferase, generating a reactive acyl glucuronide. The latter is exported from hepatocytes by the ATP-dependent drug transporter MRP2. Patients with variant alleles resulting in a lower CYP2C9 activity would be expected to shunt more diclofenac through the glucuronidation pathway. Patients with variant alleles resulting in a higher glucuronidation activity would be expected to generate more of the toxic metabolite and patients with a lower activity of the exporting pump would be expected to be exposed to higher concentrations of the toxic metabolite. In a group of 24 patients with diclofenac associated liver injury, there was no difference in the prevalence of functionally relevant CYP2C9 alleles compared to patients tolerating diclofenac, but there was indeed a higher prevalence of the UGT2B7*2 alleles conferring a higher glucuronidation activity and of a mutation in ABCC2 that may result in lower transport activity of MRP2 [60]. The ABCC2 genotype may play a role in the susceptibility to DILI from a variety of other drugs, as well as herbal medicines [61]. Glutathione transferase (GST) plays a role in the detoxification of some reactive metabolites of drugs. In a cohort of 154 patients with DILI from various drugs, carriers of double GSTT1-M1 null genotypes resulting in lower GST activity had a 2.7-fold increased risk of developing DILI compared with non-carriers [62]. The GSTM1 null genotype may also increase the risk for anti-tuberculosis drug-induced hepatotoxicity [63]. Mutations in superoxide dismutase (MnSOD) may increase the susceptibility to DILI by various drugs [63]. Polymorphisms in activating and detoxifying enzymes may thus explain, albeit to a small extent, the susceptibility to DILI.
Genetic differences, in regard to the reaction to injury, may be a risk factor for developing DILI. The genotypes of IL-10 and IL-4 exhibit a different distribution in patients experiencing diclofenac hepatotoxicity compared to patients not reacting to diclofenac [64]. Animal models suggest that an inflammatory response and in particular antiinflammatory cytokines play an important role in determining the extent of the toxicity of model compounds [65]. The HLA genotype may also be a risk factor for liver injury by certain drugs, such as diclofenac, ximelagatran and amoxicillin/clavulanic acid [66–68]. The HLA-B*5701 allele increases the risk for flucloxacillin associated cholestasis by a factor of 80 [52]. The prevalence of this allele in the population is approximately 5%. Therefore, unlike the case of abacavir hypersensitivity where genotyping may prevent toxicity [69], the value of HLA genotyping is likely to be low for predicting flucloxacillin cholostasis. Nevertheless, the observations indicate that genetic factors associated with the inflammatory response to injury and antigen presentation may play a role in idiosyncratic drug reactions.
The role of the immune response
Considering the prevalence of known polymorphisms in bioactivation pathways, detoxification reactions and transport processes and their potential effects on the exposure to toxic metabolites, it is difficult to explain the idiosyncratic nature of drug-induced hepatotoxicity: a higher incidence, a more homogenous presentation and a better predictability of DILI would be expected. The repertoire of immunological responses is more variable than the range of phenotypic manifestations of genetic differences in activating and detoxifying systems, suggesting that the immune system plays an important role in DILI (fig. 2). Since each individual shows a somewhat different response to an immunogen depending on what epitopes are recognised and what mix of effector cells are involved in the immune response, the immune system could provide a plausible explanation for the variable and rare presentation of idiosyncratic DILI.
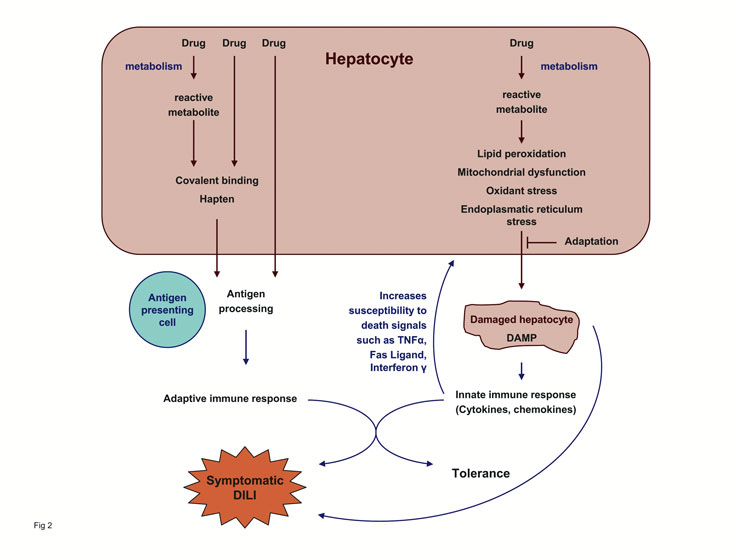
Figure 2
The concept of idiosyncratic DILI. Drug metabolism generates chemically reactive metabolites that interfere with cellular homeostasis. Unless the cell adapts to the metabolic disturbance, the hepatocyte dies and danger signals are released. The ensuing innate immune response increases the susceptibility of hepatocytes to death signals resulting in additional cell damage. Reactive metabolites bound to macromolecules, and possibly also cellular components released from damaged hepatocytes, can also elicit an adaptive immune response that may result in cell damage. The interplay between the innate immune system activated by the danger signals and the adaptive immune system may be an important factor in determining whether tolerance or symptomatic DILI ensues. Depending on the drug involved and the host response, one step or the other step may be more relevant for the outcome.
Idiosyncratic drug reactions often target the liver but may also affect other organs, where they are, in most instances, considered to reflect an immune response. The majority of drug-induced skin rashes is immune mediated [70] and most haematological drug reactions, such as haemolytic anaemia, thrombocytopenia or aplastic anemias, involve the immune system. Cases of DILI presenting with concomitant rash, fever and/or eosinophilia are likely to be immune mediated. Many of the drugs responsible for idiosyncratic DILI are also associated with autoimmune syndromes like autoimmune haemolytic anemias, lupus erythematodes, agranulocytosis, vasculitis or autoimmune hepatitis (for a list of compounds see [71]). Minocycline may serve as an example: like other tetracyclines it causes dose-dependent liver injury following large doses. With therapeutic doses, idiosyncratic hepatitis and hepatic failure associated with fever, rash, lymphadenopathy, eosinophilia and neutropenia may occur approximately one month after starting treatment [72, 73]. Later, after prolonged exposure, an auto-immune hepatitis with positive antinuclear antibodies and hypergammaglobulinemia may appear [74, 75].
The time course of idiosyncratic DILI is also suggestive of an immune mechanism. The typical interval from the start of drug exposure to the onset of DILI is – with some notable exceptions – 1–3 months, reflecting the time it takes for sensitised lymphocytes to expand in order to produce a clinically evident reaction. Although this time lag to the onset of liver injury would also be compatible with a gradual accumulation of mitochondrial damage reaching a threshold beyond which liver injury manifests itself, other features are more difficult to explain by the accumulation of mitochondrial damage. Patients with DILI usually recover promptly when the offending drug is stopped but in some cases liver damage continues, suggesting an auto-immune component. In the case of amoxicillin-clavulanic acid induced cholestatic hepatitis, it is not uncommon that the liver enzymes start to rise several weeks after the drug has been stopped [76]. A similar observation was also made with ximelagatran. Re-challenge with a drug that has caused DILI can result in rapid recurrence of liver injury. An anamnestic response mediated by memory T and B cells is typical for an immune response. On the other hand, an anamnestic response and systemic symptoms such as fever and rash are often missing. This does not favour an immune mechanism but does not necessarily argue against it.
Inter-individual differences in epitope recognition may explain why the pattern of liver injury from a single drug may vary and why some drugs cause idiosyncratic liver injury in some patients and injury to other organs, such as the bone marrow, in others as is the case with the antiepileptic drug felbamate [77]. The immune response can be directed either against the hapten (i.e., the offending drug) alone, against the drug and a fragment of the protein it is bound to, or solely against a protein fragment of the drug-protein complex that was processed for antigen presentation. The first example would represent a classical adaptive immune response against a drug. The second example could explain the organ specificity of an immune response: if the protein fragment that, together with the hapten, elicits the immune response has organ specificity, this could explain why in some cases
injury to the liver and in others injury to another organ, for example the bone marrow, ensues. The last example where the immune response is directed solely against endogenous protein fragments could explain autoimmune reactions to drugs triggered by some unknown additional factors independent of the presence of the responsible drug.
The question arises of how small molecular drugs can elicit an immune response since small molecules (<1000 molecular mass) are considered non-immunogenic. However, reactive metabolites of a drug or, less commonly, the parent drug itself, as in the case of penicillin, may act as haptens by covalently binding to proteins and forming immunogenic drug-protein adducts which, after processing by antigen-presenting cells, elicit either cytotoxic T-cell or antibody responses. One of the first examples in support of the hapten hypothesis was the anaesthetic halothane, which is in part metabolised to trifluoroacetic acid. Antibodies against trifluoroacetylated proteins are found in the sera of patients who have experienced severe hepatic injury from halothane. These antibodies together with mononuclear cells are able to lyse hepatocytes that were exposed to halothane in vitro. Antibody-dependent cell-mediated injury of diclofenac-treated mouse hepatocytes has also been shown [78]. Mice immunised with trifluoroacetylated proteins develop liver injury resembling auto-immune hepatitis [79]. Antibodies against CYP2E1 are also found in anaesthetists exposed to halothane without evidence for liver injury [80]. In contrast to patients who had an episode of halothane hepatitis where the antibodies are of the subclass IgG4, the antibodies in subjects exposed to halothane are IgG1 [81]. It has been suggested that unlike IgG1 immune complexes, IgG4-containing immune complexes escape clearance and lead to liver injury. The reactive metabolite of tycrinafen, a diuretic that has been withdrawn from the market because of its hepatotoxicity, binds to the cytochrome P450 which forms the metabolite [82]. Patients with tycrinafen hepatitis form unique antibodies, LKM2, which react with drug-modified CYP450 [83]. A reactive metabolite of the antihypertensive agent dihydralazine binds to CYP1A2 by which it is generated and results in the formation of anti-CYP1A2 antibodies in some patients [84]. The antibodies were not present in patients treated with dihydralazine without liver disease [85]. Whether these antibodies play a role in the pathogenesis of liver injury is not clear, but they demonstrate an immune response to proteins modified by reactive metabolites and the possible involvement of the adaptive immune system in some idiosyncratic drug reactions.
The hapten hypothesis can not explain idiosyncratic immunologic drug reactions occurring in the absence of demonstrable covalent binding. Covalent binding of drugs and drug metabolites is not trivial to demonstrate and the absence of evidence for covalent binding may not be evidence of its absence. Nevertheless, considering their chemical nature not all reactive metabolites are likely to bind to macromolecules and form a neo-antigen. However, covalent binding may not be a prerequisite for an immune response as some drugs have been proposed to bind reversibly to major histocompatibility complexes and T-cell receptor molecules [86]. In addition, the metabolism of a drug may result in lipid peroxidation due to the generation of free radicals, to oxidative stress via an increased generation of reactive oxygen species or to endoplasmatic reticulum stress, all of which may generate danger signals that activate the innate immune system [87].
In summary, hepatic metabolism of drugs to metabolites that result in cellular dysfunction by causing oxidative stress, mitochondrial impairment lipid peroxidation or, seems to underlie most instances of DILI. Genetic polymorphisms in activating and detoxifying enzymes determine, in part, the extent of cellular stress. In the case of paracetamol intoxication, the ensuing dose-dependent cellular damage and the host reaction to this damage results in predictable hepatic failure. In the case of idiosyncratic liver injury, the mechanisms involved and the sequence of events are more speculative but may be similar: formation of a reactive metabolite, disruption of cellular homeostasis, mitochondrial dysfunction, activation of death-promoting pathways and release of drug-modified macromolecules and/or danger signals that in turn may elicit an immune response. Depending on the drug involved, one step or another in the cascade of events may be more or less decisive for the development of DILI in the few patients who are not able to adapt to the cellular stress and contain the damage.
Correspondence:
Bernhard Lauterburg, MD
Institute for clinical pharmacology and visceral research
Murtenstrasse 35
3010 Bern
Switzerland
blauterburg@ikp.unibe.ch
References
1 Chalasani N, Fontana RJ, Bonkovsky HL, Watkins PB, Davern T, Serrano J, et al. Causes, clinical features, and outcomes from a prospective study of drug-induced liver injury in the United States. Gastroenterology. 2008;135:1924–34.
2 de Abajo FJ, Montero D, Madurga M, Garcia Rodriguez LA. Acute and clinically relevant drug-induced liver injury: a population based case-control study. Br J Clin Pharmacol. 2004;58:71–80.
3 Bell LN, Chalasani N. Epidemiology of idiosyncratic drug-induced liver injury. Semin Liver Dis. 2009;29:337–47.
4 Temple RJ, Himmel MH. Safety of newly approved drugs: implications for prescribing. JAMA. 2002;287:2273–5.
5 Lasser KE, Allen PD, Woolhandler SJ, Himmelstein DU, Wolfe SM, Bor DH. Timing of new black box warnings and withdrawals for prescription medications. JAMA. 2002;287:2215–20.
6 Biour M, Ben Salem C, Chazouilleres O, Grange JD, Serfaty L, Poupon R. [Drug-induced liver injury; fourteenth updated edition of the bibliographic database of liver injuries and related drugs]. Gastroenterol Clin Biol. 2004;28:720–59.
7 Stickel F, Patsenker E, Schuppan D. Herbal hepatotoxicity. J Hepatol. 2005;43:901–10.
8 Benichou C. Criteria of drug-induced liver disorders. Report of an international consensus meeting. J Hepatol. 1990;11:272–76.
9 Lucena MI, Andrade RJ, Kaplowitz N, Garcia-Cortes M, Fernandez MC, Romero-Gomez M, et al. Phenotypic characterization of idiosyncratic drug-induced liver injury: the influence of age and sex. Hepatology. 2009;49:2001–9.
10 Danan G, Benichou C. Causality assessment of adverse reactions to drugs – I. A novel method based on the conclusions of international consensus meetings: application to drug-induced liver injuries. J Clin Epidemiol. 1993;46:1323–30.
11 Rochon J, Protiva P, Seeff LB, Fontana RJ, Liangpunsakul S, Watkins PB, et al. Reliability of the Roussel Uclaf Causality Assessment Method for assessing causality in drug-induced liver injury. Hepatology. 2008;48:1175–83.
12 Fontana RJ, Watkins PB, Bonkovsky HL, Chalasani N, Davern T, Serrano J, et al. Drug-Induced Liver Injury Network (DILIN) prospective study: rationale, design and conduct. Drug Saf. 2009;32:55–68.
13 James LP, Letzig L, Simpson PM, Capparelli E, Roberts DW, Hinson JA et al. Pharmacokinetics of acetaminophen-protein adducts in adults with acetaminophen overdose and acute liver failure. Drug Metab Dispos. 2009;37:1779–84.
14 Bjornsson E, Olsson R. Outcome and prognostic markers in severe drug-induced liver disease. Hepatology. 2005;42:481–9.
15 Bjornsson E, Davidsdottir L. The long-term follow-up after idiosyncratic drug-induced liver injury with jaundice. J Hepatol. 2009;50:511–7.
16 Andrade RJ, Lucena MI, Fernandez MC, Pelaez G, Pachkoria K, Garcia-Ruiz E et al. Drug-induced liver injury: an analysis of 461 incidences submitted to the Spanish registry over a 10-year period. Gastroenterology. 2005;129:512–21.
17 Aithal PG, Day CP. The natural history of histologically proved drug induced liver disease. Gut. 1999;44:731–5.
18 Lammert C, Einarsson S, Saha C, Niklasson A, Bjornsson E, Chalasani N. Relationship between daily dose of oral medications and idiosyncratic drug-induced liver injury: search for signals. Hepatology. 2008;47:2003–9.
19 Russo MW, Galanko JA, Shrestha R, Fried MW, Watkins P. Liver transplantation for acute liver failure from drug induced liver injury in the United States. Liver Transpl. 2004;10:1018–23.
20 Lammert C, Bjornsson E, Niklasson A, Chalasani N. Oral medications with significant hepatic metabolism at higher risk for hepatic adverse events. Hepatology. 2009.
21 Uetrecht JP. New concepts in immunology relevant to idiosyncratic drug reactions: the “danger hypothesis” and innate immune system. Chem Res Toxicol. 1999;12:387–95.
22 Tostmann A, Boeree MJ, Aarnoutse RE, de Lange WC, van d, V, Dekhuijzen R. Antituberculosis drug-induced hepatotoxicity: concise up-to-date review. J Gastroenterol Hepatol. 2008;23:192–202.
23 Kumar R, Bhatia V, Khanal S, Sreenivas V, Gupta SD, Panda SK et al. Antituberculosis therapy-induced acute liver failure: magnitude, profile, prognosis, and predictors of outcome. Hepatology. 2010;51:1665–74.
24 Gupta NK, Lewis JH. Review article: The use of potentially hepatotoxic drugs in patients with liver disease. Aliment Pharmacol Ther. 2008;28:1021–41.
25 Russo MW, Watkins PB. Are patients with elevated liver tests at increased risk of drug-induced liver injury? Gastroenterology. 2004;126:1477–80.
26 Nierkens S, Pieters R. Murine models of drug hypersensitivity. Curr Opin Allergy Clin Immunol. 2005;5:331–5.
27 Ong MM, Latchoumycandane C, Boelsterli UA. Troglitazone-induced hepatic necrosis in an animal model of silent genetic mitochondrial abnormalities. Toxicol Sci. 2007;97:205–13.
28 Kashimshetty R, Desai VG, Kale VM, Lee T, Moland CL, Branham WS et al. Underlying mitochondrial dysfunction triggers flutamide-induced oxidative liver injury in a mouse model of idiosyncratic drug toxicity. Toxicol Appl Pharmacol. 2009;238:150–9.
29 Mitchell JR, Jollow DJ, Potter WZ, Davis DC, Gillette JR, Bro-die BB. Acetaminophen-Induced Hepatic Necrosis .1. Role of Drug-Metabolism. J Pharmacol Exp Ther. 1973;187:185–94.
30 Lauterburg BH, Corcoran GB, Mitchell JR. Mechanism of action of N-acetylcysteine in the protection against the hepatotoxicity of acetaminophen in rats in vivo. J Clin Invest. 1983;71:980–91.
31 Lauterburg BH, Velez ME. Glutathione deficiency in alcoholics: risk factor for paracetamol hepatotoxicity. Gut. 1988;29:1153–7.
32 Seeff LB, Cuccherini BA, Zimmerman HJ, Adler E, Benjamin SB. Acetaminophen hepatotoxicity in alcoholics. A therapeutic misadventure. Ann Intern Med. 1986;104:399–404.
33 Tirmenstein MA, Nelson SD. Subcellular binding and effects on calcium homeostasis produced by acetaminophen and a nonhepatotoxic regioisomer, 3‘-hydroxyacetanilide, in mouse liver. J Biol Chem. 1989;264:9814–9.
34 Griffith OW, Meister A. Origin and turnover of mitochondrial glutathione. Proc Natl Acad Sci. USA. 1985;82:4668–72.
35 Gunawan BK, Liu ZX, Han D, Hanawa N, Gaarde WA, Kaplowitz N. c-Jun N-terminal kinase plays a major role in murine acetaminophen hepatotoxicity. Gastroenterology. 2006;131:165–78.
36 Latchoumycandane C, Goh CW, Ong MMK, Boelsterli UA. Mitochondrial protection by the JNK inhibitor leflunomide rescues mice from acetaminophen-induced liver injury. Hepatology. 2007;45:412–21.
37 Henderson NC, Pollock KJ, Frew J, Mackinnon AC, Flavell RA, Davis RJ, et al. Critical role of c-jun (NH2) terminal kinase in paracetamol- induced acute liver failure. Gut. 2007;56:982–90.
38 Chen CJ, Kono H, Golenbock D, Reed G, Akira S, Rock KL. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med. 2007;13:851–6.
39 Imaeda AB, Watanabe A, Sohail MA, Mahmood S, Mohamadnejad M, Sutterwala FS, et al. Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J Clin Invest. 2009;119:305–14.
40 Wallace KB, Starkov AA. Mitochondrial targets of drug toxicity. Annual Review of Pharmacology and Toxicology. 2000;40:353–88.
41 Boelsterli UA, Lim PL. Mitochondrial abnormalities--a link to idiosyncratic drug hepatotoxicity? Toxicol Appl Pharmacol. 2007;220:92–107.
42 McKenzie R, Fried MW, Sallie R, Conjeevaram H, Di Bisceglie AM, Park Y, et al. Hepatic failure and lactic acidosis due to fialuridine (FIAU), an investigational nucleoside analogue for chronic hepatitis B. N Engl J Med. 1995;333:1099–105.
43 Krahenbuhl S, Brandner S, Kleinle S, Liechti S, Straumann D. Mitochondrial diseases represent a risk factor for valproate-induced fulminant liver failure. Liver. 2000;20:346–8.
44 Kopanoff DE, Snider DE Jr, Caras GJ. Isoniazid-related hepatitis: a U.S. Public Health Service cooperative surveillance study. Am Rev Respir Dis. 1978;117:991–1001.
45 Watkins PB, Whitcomb RW. Hepatic dysfunction associated with troglitazone. N Engl J Med. 1998;338:916–7.
46 Watkins PB, Zimmerman HJ, Knapp MJ, Gracon SI, Lewis KW. Hepatotoxic effects of tacrine administration in patients with Alzheimer‘s disease. JAMA. 1994;271:992–8.
47 Lee WM, Larrey D, Olsson R, Lewis JH, Keisu M, Auclert L, et al. Hepatic findings in long-term clinical trials of ximelagatran. Drug Saf. 2005;28:351–70.
48 Ros JE, Libbrecht L, Geuken M, Jansen PL, Roskams TA. High expression of MDR1, MRP1, and MRP3 in the hepatic progenitor cell compartment and hepatocytes in severe human liver disease. J Pathol. 2003;200:553–60.
49 Aleksunes LM, Campion SN, Goedken MJ, Manautou JE. Acquired resistance to acetaminophen hepatotoxicity is associated with induction of multidrug resistance-associated protein 4 (Mrp4) in proliferating hepatocytes. Toxicol Sci. 2008;104:261–73.
50 Shayiq RM, Roberts DW, Rothstein K, Snawder JE, Benson W, Ma X, et al. Repeat exposure to incremental doses of acetaminophen provides protection against acetaminophen-induced lethality in mice: an explanation for high acetaminophen dosage in humans without hepatic injury. Hepatology. 1999;29:451–63.
51 Eksteen B, Afford SC, Wigmore SJ, Holt AP, Adams DH. Immune-mediated liver injury. Semin Liver Dis. 2007;27:351–66.
52 Daly AK, Donaldson PT, Bhatnagar P, Shen Y, Pe’er I, Floratos A, et al. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat Genet. 2009;41:816–9.
53 Fukino K, Sasaki Y, Hirai S, Nakamura T, Hashimoto M, Yamagishi F, et al. Effects of N-acetyltransferase 2 (NAT2), CYP2E1 and Glutathione-S-transferase (GST) genotypes on the serum concentrations of isoniazid and metabolites in tuberculosis patients. J Toxicol Sci. 2008;33:187–95.
54 Peretti E, Karlaganis G, Lauterburg BH. Increased urinary excretion of toxic hydrazino metabolites of isoniazid by slow acetylators. Effect of a slow-release preparation of isoniazid. Eur J Clin Pharmacol. 1987;33:283–6.
55 Dickinson DS, Bailey WC, Hirschowitz BI, Soong SJ, Eidus L, Hodgkin MM. Risk factors for isoniazid (NIH)-induced liver dysfunction. J Clin Gastroenterol. 1981;3:271–9.
56 Huang YS, Chern HD, Su WJ, Wu JC, Lai SL, Yang SY, et al. Polymorphism of the N-acetyltransferase 2 gene as a susceptibility risk factor for antituberculosis drug-induced hepatitis. Hepatology. 2002;35:883–9.
57 Possuelo LG, Castelan JA, de Brito TC, Ribeiro AW, Cafrune PI, Picon PD, et al. Association of slow N-acetyltransferase 2 profile and anti-TB drug-induced hepatotoxicity in patients from Southern Brazil. Eur J Clin Pharmacol. 2008;64:673–81.
58 Huang YS, Chern HD, Su WJ, Wu JC, Chang SC, Chiang CH, et al. Cytochrome P450 2E1 genotype and the susceptibility to antituberculosis drug-induced hepatitis. Hepatology. 2003;37:924–30.
59 Cho HJ, Koh WJ, Ryu YJ, Ki CS, Nam MH, Kim JW et al. Genetic polymorphisms of NAT2 and CYP2E1 associated with antituberculosis drug-induced hepatotoxicity in Korean patients with pulmonary tuberculosis. Tuberculosis. (Edinb ). 2007;87:551–6.
60 Daly AK, Aithal GP, Leathart JB, Swainsbury RA, Dang TS, Day CP. Genetic susceptibility to diclofenac-induced hepatotoxicity: contribution of UGT2B7, CYP2C8, and ABCC2 genotypes. Gastroenterology. 2007;132:272–81.
61 Choi JH, Ahn BM, Yi J, Lee JH, Lee JH, Nam SW, et al. MRP2 haplotypes confer differential susceptibility to toxic liver injury. Pharmacogenet Genomics. 2007;17:403–15.
62 Lucena MI, Andrade RJ, Martinez C, Ulzurrun E, Garcia-Martin E, Borraz Y, et al. Glutathione S-transferase m1 and t1 null genotypes increase susceptibility to idiosyncratic drug-induced liver injury. Hepatology. 2008;48:588–96.
63 Huang YS, Su WJ, Huang YH, Chen CY, Chang FY, Lin HC, et al. Genetic polymorphisms of manganese superoxide dismutase, NAD(P)H:quinone oxidoreductase, glutathione S-transferase M1 and T1, and the susceptibility to drug-induced liver injury. J Hepatol. 2007;47:128–34.
64 Aithal GP, Ramsay L, Daly AK, Sonchit N, Leathart JB, Alexander G, et al. Hepatic adducts, circulating antibodies, and cytokine polymorphisms in patients with diclofenac hepatotoxicity. Hepatology. 2004;39:1430–40.
65 Bourdi M, Eiras DP, Holt MP, Webster MR, Reilly TP, Welch KD, et al. Role of IL-6 in an IL-10 and IL-4 double knockout mouse model uniquely susceptible to acetaminophen-induced liver injury. Chem Res Toxicol. 2007;20:208–16.
66 Kindmark A, Jawaid A, Harbron CG, Barratt BJ, Bengtsson OF, Andersson TB, et al. Genome-wide pharmacogenetic investigation of a hepatic adverse event without clinical signs of immunopathology suggests an underlying immune pathogenesis. Pharmacogenomics J. 2008;8:186–95.
67 Hautekeete ML, Horsmans Y, Van WC, Demanet C, Henrion J, Verbist L, et al. HLA association of amoxicillin-clavulanate-induced hepatitis. Gastroenterology. 1999;117:1181–6.
68 O’Donohue J, Oien KA, Donaldson P, Underhill J, Clare M, MacSween RN, et al. Co-amoxiclav jaundice: clinical and histological features and HLA class II association. Gut. 2000;47:717–20.
69 Mallal S, Phillips E, Carosi G, Molina JM, Workman C, Tomazic J, et al. HLA-B*5701 screening for hypersensitivity to abacavir. N Engl J Med. 2008;358:568–79.
70 Lerch M, Pichler WJ. The immunological and clinical spectrum of delayed drug-induced exanthems. Curr Opin Allergy Clin Immunol. 2004;4:411–9.
71 Uetrecht J. Immunoallergic drug-induced liver injury in humans. Semin Liver Dis. 2009;29:383–92.
72 Knowles SR, Shapiro L, Shear NH. Serious adverse reactions induced by minocycline. Report of 13 patients and review of the literature. Arch Dermatol. 1996;132:934–9.
73 Kaufmann D, Pichler W, Beer JH. Severe episode of high fever with rash, lymphadenopathy, neutropenia, and eosinophilia after minocycline therapy for acne. Arch Intern Med. 1994;154:1983–4.
74 Gough A, Chapman S, Wagstaff K, Emery P, Elias E. Minocycline induced autoimmune hepatitis and systemic lupus erythematosus-like syndrome. BMJ. 1996;312:169–72.
75 Goldstein NS, Bayati N, Silverman AL, Gordon SC. Minocycline as a cause of drug-induced autoimmune hepatitis. Report of four cases and comparison with autoimmune hepatitis. Am J Clin Pathol. 2000;114:591–8.
76 Mari JY, Guy C, Beyens MN, Ollagnier M. Delayed drug-induced hepatic injury. Evoking the role of amoxicillin-clavulinic acid combination. Therapie. 2000;55:699–704.
77 Pellock JM. Felbamate. Epilepsia. 1999;40(Suppl 5):S57–62.
78 Kretz-Rommel A, Boelsterli UA. Cytotoxic activity of T cells and non-T cells from diclofenac-immunized mice against cultured syngeneic hepatocytes exposed to diclofenac. Hepatology. 1995;22:213–22.
79 Njoku DB, Talor MV, Fairweather D, Frisancho-Kiss S, Odumade OA, Rose NR. A novel model of drug hapten-induced hepatitis with increased mast cells in the BALB/c mouse. Exp Mol Pathol. 2005;78:87–100.
80 Njoku DB, Greenberg RS, Bourdi M, Borkowf CB, Dake EM, Martin JL, et al. Autoantibodies associated with volatile anesthetic hepatitis found in the sera of a large cohort of pediatric anesthesiologists. Anesth Analg. 2002;94:243–9, table.
81 Njoku DB, Mellerson JL, Talor MV, Kerr DR, Faraday NR, Outschoorn I, et al. Role of CYP2E1 immunoglobulin G4 subclass antibodies and complement in pathogenesis of idiosyncratic drug-induced hepatitis. Clin Vaccine Immunol. 2006;13:258–65.
82 Robin MA, Maratrat M, Le RM, Le Breton FP, Bonierbale E, Dansette P, et al. Antigenic targets in tienilic acid hepatitis. Both cytochrome P450 2C11 and 2C11-tienilic acid adducts are transported to the plasma membrane of rat hepatocytes and recognized by human sera. J Clin Invest. 1996;98:1471–80.83 Lecoeur S, Andre C, Beaune PH. Tienilic acid-induced autoimmune hepatitis: anti-liver and-kidney microsomal type 2 autoantibodies recognize a three-site conformational epitope on cytochrome P4502C9. Mol Pharmacol. 1996;50:326–33.
84 Bourdi M, Tinel M, Beaune PH, Pessayre D. Interactions of dihydralazine with cytochromes P4501A: a possible explanation for the appearance of anti-cytochrome P4501A2 autoantibodies. Mol Pharmacol. 1994;45:1287–95.
85 Bourdi M, Larrey D, Nataf J, Bernuau J, Pessayre D, Iwasaki M, et al. Anti-liver endoplasmic reticulum autoantibodies are directed against human cytochrome P-450IA2. A specific marker of dihydralazine-induced hepatitis. J Clin Invest. 1990;85:1967–73.
86 Pichler WJ, Beeler A, Keller M, Lerch M, Posadas S, Schmid D, et al. Pharmacological interaction of drugs with immune receptors: the p-i concept. Allergol Int. 2006;55:17–25.
87 Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045.:991–1045.