Insights into molecular pathways for targeted therapeutics in acute leukaemia
DOI: https://doi.org/10.4414/smw.2010.13068
V
Stavropoulou, L
Brault, J
Schwaller
Summary
Despite the development of modern chemotherapeutic regimens, acute leukaemia remains incurable in the majority of adult patients and potential cure is associated with considerable side effects. Clinical and experimental research of the last two decades has demonstrated that acute leukaemia is the consequence of multiple collaborative molecular aberrations affecting protein kinases and transcriptional regulators induced by genetic alterations and/or epigenetic mechanisms. New technologies have been developed to detect aberrations of the entire (epi)genome of a leukaemic blast that will result in a long list of potential therapeutic targets needing to be functionally validated in cellular and animal leukaemia models. Using these methods, several "druggable” protein kinases have been identified. These kinases exert their oncogenic potential not only through expansion of the leukaemic clone, but also by regulating critical interactions of leukaemic stem cells with the microenvironment. Due to the molecular complexity of acute leukaemia, new functional genome-wide screens have been established and may help to identify targets that when blocked result in synthetic lethality of the leukaemic blasts harbouring distinct (epi)genomic lesions. A close interaction between the academic and the pharmaceutical biomedical research will be essential to translate these exciting new molecular findings into improved therapies for acute leukaemia.
Introduction
Acute leukaemia is a disorder of the haematopoietic system characterised by the expansion of a clonal population of cells that is blocked in differentiation. In paediatric patients, acute leukaemia more frequently involves the lymphoid lineage leading to acute lymphoblastic leukaemia (ALL), whereas in older patients the myeloid lineage is predominantly affected, leading to acute myeloid leukaemia (AML). Current treatment regimens using combinatory chemotherapy result in long-term survival rates in more than 80% of paediatric ALL patients, whereas less than half of paediatric AML or infant leukaemia can be cured [1]. Although standard chemotherapy induces remission rates of 50–80% for adult AML patients, the majority of patients relapse, resulting in high mortality rates [2]. New treatment strategies are therefore needed not only to improve the cure rate but also the patients’ quality of life. The presence of distinct cytogenetic lesions is closely linked to prognosis. Modern molecular biology tools have resulted in a better understanding of the disease’s pathogenesis. A large number of genetic alterations have been found and their leukaemogenic potential has been characterised in various in vitro and in vivo experimental systems. Following the success of small molecule inhibitors such as imatinib-mesylate, the targeting of aberrant protein kinase activity that is frequently associated with chronic myeloid leukaemia has raised strong hopes that modern molecular genetics will also help to develop novel therapies for patients with acute leukaemia. Here, we provide a short overview of current understanding regarding the functionally interfering genetic alterations that lead to acute leukaemia. In addition, we summarise the most recent efforts (including our own) to identify potential new therapeutic approaches targeting not only the leukaemic blast but also its interaction with the microenvironment.
Genetics of acute leukaemia
Known genetic alterations
Biologically, acute leukaemia is characterised by uncontrolled accumulation of haematopoietic progenitor cells with a blocked differentiation programme. Intensive research in the last two decades has provided convincing evidence that acute leukaemia is the product of a multistep process associated with the acquisition of multiple genetic alterations that have been functionally classified into two major categories (fig. 1, Sup. Table 1) [3].
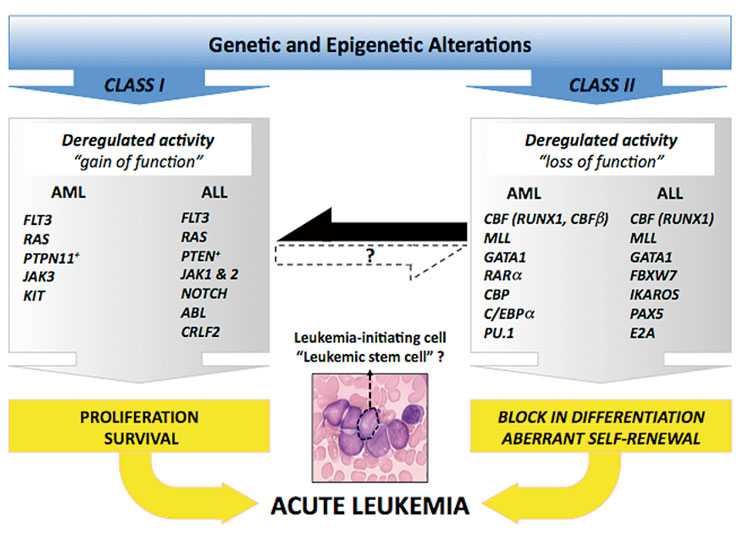
Figure 1
(Epi)genomic alterations in acute leukaemia.
Aberrant "class I” activities lead to proliferation and survival advantage of the cells whereas "class II” modifications impose a block in differentiation process and result in aberrant self-renewal. The black and dashed arrows represent the existing and putative crosstalks between the different class mutations respectively.
Uncontrolled activation of a large number of signalling mediators that regulate proliferation and/or survival is the consequence of a gain of function mutations or deregulated expression, and is often referred as "class I mutations”. The best-studied examples are protein tyrosine kinases (such as FLT3, KIT or JAK), RAS family members or the RAF/MEK/ERK and PI3K/AKT signalling pathways [4]. Somatic mutations leading to constitutive activation of FLT3 (internal tandem duplications, ITDs; or kinase domain point mutations) are found in up to 30% of adult AML patients [4]. RAS activation in acute leukaemia is caused by either enhanced signalling of upstream mediators such as FLT3, or through somatic mutations which lead to amino acid changes in codons 12, 13, or 61. This results in its constitutive activation. "Gain of function” mutations in N-RAS and K-RAS (rarely in H-RAS) are found in 5–10% of adult AML patients [4]. In contrast to AML, acute lymphoblastic leukaemia (ALL) is characterised by alterations of the ABL gene such as BCR/ABL fusions in B-cell ALL or NUP214/ABL in T-cell ALL [5]. Gain of function mutations (point mutations or fusions) of the NOTCH1 gene are among the most frequent alterations in T-ALL. Aberrant NOTCH1 activity seems to support the proliferation and survival of T-cell precursors by activating several downstream mediators such as the NF-κB or the PI3K/AKT pathway [6].
Impaired differentiation and aberrant self-renewal capacity is predominantly the consequence of alterations of genes encoding transcriptional regulators, and they are often categorised as "class II” mutations. These genes include factors that ensure the enrollment of a genetic program controlling the formation of all terminal blood cell lineages. In AML the core-binding factor (CBF), the retinoid acid receptor-alpha (RARα), or the mixed-lineage leukaemia 1 (MLL1) genes are targets of a large number of chromosomal translocations leading to chimeric fusion proteins with aberrant transcriptional activity. More than 20% of adult AML-M2 cases are characterised by the presence of a translocation involving CBF components such as t(8;21) or inv16 leading to AML1/ETO or CBFβ/MYH11 respectively. RARα fusion genes are the molecular correlates of acute promyelocytic leukaemia (APL). The MLL1 gene on chromosome 11q23 is involved in translocations resulting in fusions with more than 50 different partners. The translocations t(4;11), t(9;11) and t(11;19) leading to MLL/AF4, MLL/AF9 and MLL/ENL account for over 60% of leukaemia-associated MLL fusions [7]. Alterations of MLL are a hallmark of acute leukaemia in infants (<1 year) and are generally considered as a poor prognostic marker [7]. Similarly, the nucleopore protein NUP98 can be fused to different homeo box transcriptional regulators or other partners, and the presence of a NUP98/HOX fusion seems to be associated with early chemoresistance and relapse of the disease [8].
Clinical and experimental studies are continuously providing new clues to the molecular mechanisms of acute leukaemia. Although conceptually correct, it is becoming evident that these interactions are much more complex than what was initially proposed as a simplified collaboration model of two classes of mutations. A class II mutation such as an MLL fusion gene can trigger the upregulation of class I targets (like the FLT3 kinase), and thus can functionally imitate the alterations occurring in both classes [9]. Likewise, downstream transcriptional executors of class I activities may also affect aberrant self-renewal that has been assigned to class II mutations. Although it is currently not known how many aberrant class I and/or class II mutations are in fact necessary for acute leukaemia to develop in humans, the existing mouse models and clinical observations indicate that certain alterations such as MLL fusions may be more independent of cooperating events as compared to others such as the CBF fusions. On the basis of these observations we propose a potential crosstalk model for acute leukaemia with emphasis on collaborative activities rather than mutations (fig. 1).
Genome-wide search for new genetic alterations
Several recent studies using genome-wide single-nucleotide polymorphism (SNP)-arrays have identified multiple novel (not detected by conventional cytogenetics) structural genetic alterations targeting key signalling pathways in leukaemic cells [10]. Analysis of paediatric ALL patients revealed unknown recurrent alterations in major regulators of B-cell development including PAX5, E2A, IKZF1 or IKZF2 [10]. Interestingly, deletions of IKZF1 (also known as IKAROS) were found not only in >80% of Philadelphia-chromosome (BCR/ABL fusion gene) positive B-cell ALL, but also as acquired lesions upon progression from CML to ALL. IKFZ1 deletions result in haploinsufficiency, expression of dominant-negative acting isoforms or complete loss of expression. The presence of IKZF1 alterations seems to be a strong prognostic factor for B-cell progenitor ALL with very poor outcome [10]. Moreover, comparative analysis of copy number changes demonstrated that B-cell ALL cells at relapse lacked some alterations present at diagnosis suggesting that the cells responsible for relapse might be present as minor subpopulations at the moment of the diagnosis of the disease. SNP-array analysis also revealed that unlike ALL, a much lower number of genetic alterations are present in paediatric AML. However, over 40% of the genomes of AML blasts from adult patients contained structural alterations not detected by conventional cytogenetic analysis [11].
In addition to the identification of somatic alterations, recent studies performing genome-wide analysis in acute lymphoblastic leukaemia (ALL) patient samples have also provided a glimpse of the possible role of inherited alleles in the pathogenesis of the disease. Genome-wide SNP-array based studies identified germline alleles possibly relevant for the molecular pathogenesis and heterogeneity of paediatric ALL. In these studies the most significant association was shown by SNPs within and near the genes encoding for IKFZ1, ARID5B and CEBPE. Future studies will be needed to demonstrate whether the germline IKFZ1 risk alleles will increase the risk of acquiring somatic alterations in this locus. In addition, the biological significance of ARID5B or CEBPE risk alleles remains to be determined [12, 13].
In their pioneer studies, Tim Ley and coworkers used massively parallel sequence analysis to compare over 95% of a primary, cytogenetically normal de novo genome for AML (FAB-M1) to a matched normal skin genome [14]. Within the coding region they identified and validated somatic gene mutations and alterations in conserved or potential regulatory elements of the genome. These mutations were heterozygous and present in almost all cells of a given leukaemia sample. In addition to mutations known to be recurrently associated with AML targeting genes such as nucleophosmin or N-RAS, they also identified a recurrent mutation in the gene coding for isocitrate-dehydrogenase (IDH1). IDH1 loss of function mutations have recently been found in over 80% of human gliomas, suggesting that aberrant IDH1 activity may play a more general role in tumourigenesis. Given the fact that about 750 point mutations were detected in the first 2 AML patients, it is very unlikely that, for example, 1 out of 100 patients would carry an identical mutation [14]. Therefore, despite the power of next-generation sequencing platforms to identify new alterations, it is necessary to analyse a significant number of additional leukaemia genome sequences to fully understand the complexity of the disease.
Learning from signalling pathways
Characterising signalling pathways unravels potential therapeutic targets
In a simplified model acute leukaemia could be proposed as the result of deregulated activity of (at least) two independent pathways. At first, an aberrant gene expression programme (mediated by altered transcriptional regulators such as a class II mutation) leads to a block in differentiation. Secondly, uncontrolled activation of a complex signal transduction network (mediated by overexpression or mutation of protein kinases and downstream targets such as a class I mutation) enhances the proliferation and/or survival of the leukaemic clone [3–5]. On the basis of this model an optimised targeted therapeutic approach would include a combination of compounds that impairs both pathways. If the functional complementation model were correct, the application of a protein tyrosine kinase inhibitor as a single drug would not be sufficient to eradicate the leukaemic blasts.
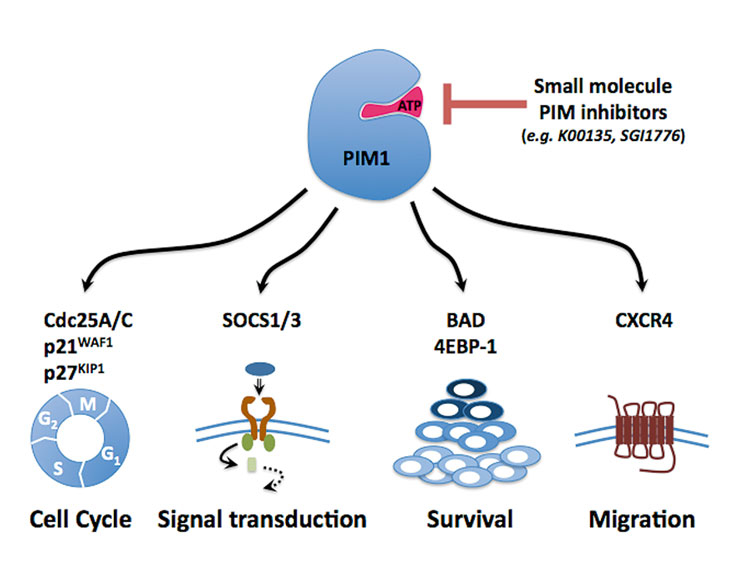
Figure 2
PIM1 as potential therapeutic target in acute leukaemia.
Different signalling pathways involving substrates of PIM1 kinase contribute to its oncogenic activity. By targeting PIM kinase with small molecule inhibitors it is not only possible to block cell cycle progression and survival of the leukaemic cells, but this may also help to mobilise the leukaemic stem cells by interfering with the CXCL12/CXCR4 axis [30].
Indeed, several clinical trials have shown that FLT3 kinase inhibitors given as a single agent for AML patients typically showed a limited and only transient biological response [15]. Several current trials are therefore testing combination of FLT3 inhibitors with standard chemotherapy. Further characterisation of the dynamic and complex network of interacting pathways downstream of a mutated signal transducer will unravel more critical nodes, which may increase the efficacy of a targeted therapeutic regimen.
Moreover, inhibition of aberrant NOTCH signalling with gamma secretase inhibitors (GSIs) shows only limited anti-leukaemic activity in human T-ALL. Activation of the PI3K-AKT pathway by NOTCH1 (or by mutation of PTEN) seems to shift the requirement of sustaining cell growth and survival from NOTCH1 to PI3K/AKT [16]. Interestingly, NOTCH1 also appears to restore glucocorticoid sensitivity of T-ALL cells while reducing GSI-mediated gut toxicity, suggesting that a combination of GSIs, PI3K/AKT inhibitors and glucocorticosteroids could be an efficient molecularly targeted regimen against T-cell ALL [17]. Transcription factor complexes such as NOTCH often lack surface involutions that would be suitable for high-affinity interaction with small molecules. However, Moellering and coworkers recently demonstrated the direct inhibition of the NOTCH transcription factor complex with a stabilised peptide that caused potent and specific antiproliferative effects in cultured cells and in a mouse model of NOTCH1-driven T-ALL [18]. This proof of principle study may pave the way for new approaches to therapeutic targeting of aberrant transcription by small molecules.
The complex signalling networks make it highly probable that a large number of downstream mediators are aberrantly activated in leukaemic blasts. At present, there is evidence that the PI3K/AKT, NF-κB or WNT pathways, for example, are inappropriately activated and associated with the maintenance of the malignant state [3–5]. In consequence, new small molecules (such as the mTOR inhibitors rapamycin and derivates) targeting one or more critical mediators of these pathways have been shown to impair the growth and/or survival of leukaemic blasts and have entered clinical trials. However, further studies are needed to understand the dynamic interplay of signalling pathways that are critical for the induction and maintenance of a malignant phenotype during diagnosis, remission and relapse of the disease.
PIM kinases as potential new therapeutic targets
The signal transducer and activator of transcription 5 (STAT5) is a central mediator of multiple upstream genetically altered protein kinases, and constitutive activation of STAT5 is a hallmark of a large proportion of haematological malignancies [19]. Using a genetic approach in a mouse model, we have previously demonstrated that STAT5 is essential for induction of a lethal myelo/lymphoproliferative disorder by a leukaemia-associated TEL/JAK2 tyrosine kinase fusion [20]. Constitutive activation of STAT5 leads to the expression of a large number of downstream targets, including PIM1, a member of three protein serine/threonine kinases (PIM1, PIM2, PIM3) that have been identified by retroviral gene tagging in a mouse lymphoma model [21].
PIMs are ubiquitously expressed and constitutive active serine/threonine kinases chiefly regulated at the transcriptional and post-translational level. Increased PIM kinase expression can act as a collaborating oncogenic event, as shown in several transgenic mouse models. PIMs seem to exert their oncogenic potential through several substrates supporting cellular proliferation, survival and migration [22] (fig. 2). Overexpression of PIM1 and PIM2 has been found in haematological malignancies and in some solid cancers (e.g., prostate carcinoma) [23]. We have previously shown that transformation of haematopoietic cells by constitutive active protein tyrosine kinases (such as the BCR/ABL fusion or the FLT3-ITD mutation) leads to significant upregulation of PIM1 and PIM2 expression [24]. Interestingly, PIM1 seems also to be regulated by HOX transcription factors known to be essential mediators of leukaemogenic class II mutations such as MLL and NUP98-fusion genes [25]. Using a genetic approach, we demonstrated that PIM1 and PIM2 are important for the growth and survival of transformed haematopoietic cells, suggesting that blocking of PIM kinases may have anti-leukaemic potential [24].
Solving the PIM1 crystal structure facilitated the development of small molecule PIM inhibitors. In collaboration with structural chemists we characterised a group of small molecules (imidazo[1,2]pyridazines) that specifically interact with and inhibit PIM kinases with low nanomolar potency. The high-resolution crystal structure of the complex PIM1/imidazo[1,2]pyridazine revealed that these inhibitors are ATP- competitive but not ATP-mimetic compounds, explaining their enhanced selectivity with respect to conventional kinase inhibitors [26]. Furthermore, the specificity of 156 validated kinase inhibitors was further tested against 60 human serine/threonine kinases. This analysis revealed many unexpected cross-reactivities for inhibitors that were thought to be specific for certain targets. For example, LY333 531, a PKCbeta inhibitor proposed for the treatment of diabetic complications, also efficiently inhibited PIM1 [27]. Both the imidazo[1,2]pyridazines and LY333 531 impaired the survival and clonogenic growth of a panel of human acute leukaemia cells, and suppressed the in vitro growth of leukaemic blasts from five AML patients but not the growth of normal umbilical cord blood mononuclear cells. The antileukaemic activity of these compounds seems to be mediated at least in part by the inhibition of PIM kinases, as shown by in vitro kinase assays and immunoblotting showing inhibition of the phosphorylation of known PIM substrates [26, 27]. A structurally related imidazo[1,2]pyridazine PIM inhibitor (SGI-1776) has recently entered the first round of clinical trials for the treatment of haematological and solid cancers [28].
Leukaemic disorders, induced by well-defined genetic alterations such as constitutive active kinases and/or transcription factor fusions, can be modelled in the mouse. By expressing the oncogene of interest in the haematopoietic system, we and other groups have shown that expression of constitutively active PTK fusions or mutants (such as X-ABL, X-JAK2 or FLT3-ITD) results in the induction of a myeloproliferative disease in mice that models the human disease in many aspects [29]. Using this approach, we addressed the role of PIM kinases for the development of a myeloproliferative disease induced by the oncogenic FLT3-ITD mutant, since previous in vitro studies have proposed that PIM2 rather than PIM1 is essential for the proliferation and survival of FLT3-ITD transformed haematopoietic cells [24].
Interestingly, whereas transplantation of FLT3-ITD expressing bone marrow cells from wildtype or PIM2–/– mice induced a typical fatal myeloproliferation, no animals reconstituted with FLT3-ITD expressing PIM1–/– developed disease. Reconstitution experiments revealed that PIM1–/– bone marrow cells are impaired in grafting the recipient animals [30]. Homing, trans-marrow migration and lodging process leading to the engraftment of haematopoietic stem cells are molecularly not very well understood. This process seems to be guided by several ligand/receptor interactions involving integrins, CXCR4 and other chemokine receptors [31]. Surprisingly, we observed that bone marrow cells from PIM1–/– mice expressed less CXCR4 at their surface than cells from PIM2–/– or wildtype animals, and were impaired in CXCL12-induced signalling. By using different approaches, we found that PIM1 activity is essential for proper CXCR4 surface expression and migration towards a CXCL12 gradient. Moreover, we found elevated PIM1 expression levels in primary AML blasts which correlated with high levels of surface CXCR4 that could be significantly reduced by treatment with a PIM1 inhibitor [30]. This work not only revealed that PIM1 plays an important role in the homing and migration of the haematopoietic cells but also suggests that small molecule PIM inhibitors might function both by inhibiting the proliferation and survival of the leukaemic cells but also by interfering with the interactions of the leukaemic cells with their microenvironment.
Therapeutic targeting of the leukaemic microenvironment
Normal blood formation is tightly regulated by interactions of the haematopoietic stem and progenitor cells with the bone marrow stroma (composed of osteoblasts, osteoclasts, endothelial cells and mesenchymal stem cells) through the production of cytokines, chemokines and multiple adhesion-initiated cellular signals [32]. The leukaemic cells can take advantage of the protective and supportive features of the microenvironment in the bone marrow to enhance their growth and survival. In addition, they may also influence inflammatory signalling to suppress the immune response. Hence targeting of the interactions of leukaemic cells with the microenvironment may repress the benefit from the niche and restore the endogenous inhibitory pathways (fig. 3). One of the best-studied mediators is the stroma-produced CXCL12 chemokine that binds to CXCR4, a member of the seven transmembrane G-coupled protein receptors expressed in HSC and progenitors [33]. Several studies have shown that CXCL12/CXCR4 signalling plays an essential role in the homing and maintenance of normal haematopoietic stem cells [33]. Most leukaemic blasts from ALL and AML patients express CXCR4, and elevated surface expression of this receptor has been proposed as a negative prognostic marker for AML patients. Functional studies have provided evidence that the CXCL12/CXCR4 signalling axis apparently facilitates the escape of malignant stem cells from chemotherapy by retention in the bone marrow microenvironment, favouring relapse of the disease. Blocking CXCR4 with small molecule antagonists (like AMD3100 [Plerixafor] or AMD3465) can disrupt tumour-stroma interactions and mobilise the leukaemia cells from the protective microenvironment [34]. In vivo studies using a transgenic mouse leukaemia model demonstrated that chemotherapy and the CXCR4 antagonist resulted in a significantly decreased tumour burden and improved overall survival [35]. These observations suggest that targeting of the leukaemic stroma by interfering with the CXCL12/CXCR4 axis may offer a novel antileukaemic therapeutic approach which is the subject of several ongoing clinical trials [34].
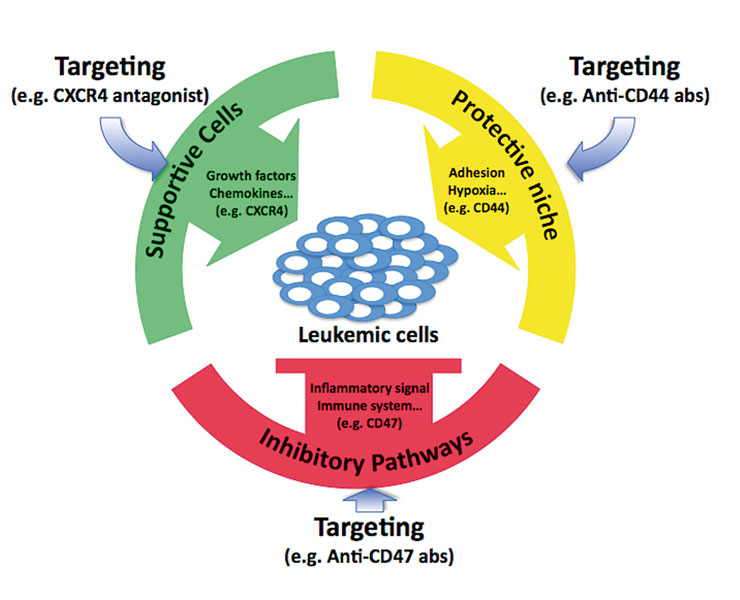
Figure 3
Potential therapeutic targeting of interactions of leukaemic cells with the micro-environment.
In the bone marrow, leukaemic cells can take advantage of the protective and supportive feature of the microenvironment, and may also impair inflammatory signal and immune response. Targeting the interactions with the microenvironment may repress the benefit from the niche and restore the endogenous inhibitory pathways to control the leukaemic cell burden.
Aberrant expression of chemokines and their receptors appears to play an important role in the biology of acute leukaemia. By gene expression profiling in a mouse model of T-cell ALL, Aifantis and colleagues recently demonstrated that the chemo-kine receptor CCR7 is a critical adhesion signal required for the targeting of leukaemic T-cells into the central nervous system (CNS). Their studies suggest that targeting the stroma, by blocking the CCL19/CCR7 axis, will reduce the risk of invasion of the CNS and subsequently relapse of the disease [36].
Like normal haematopoietic stem cells, leukaemic cells seem also to reside within specialised areas of the bone marrow that are generally called the niches. Functional studies have proposed the presence of two niches: the so-called "osteoblastic niche”, localised at the inner bone surface chiefly composed of osteoblasts, osteoclasts and stromal fibroblasts, which appear to provide an ideal environment for slowly growing long-term HSCs; and the "vascular niche”, consisting of endothelial cells lining the blood vessels, which appear to promote the proliferation and differentiation of the dividing short-term HSCs and progenitor cells [37]. An increasing number of ligand/receptor interactions have been characterised as essential regulators of proliferation and/or self-renewal of the normal and malignant haematopoietic stem cells [38]. Thus the microenvironment will not only offer protection and support to normal but also to leukaemic cells and should, therefore, be considered a potential therapeutic target (fig. 3).
Over a decade ago, John Dick and coworkers provided the first convincing evidence that similarly to normal haematopoiesis, human acute leukaemia is also characterised by a cellular hierarchy with a pool of leukaemic stem cells (LSCs) that are able to propagate the disease in a xenograft model [39]. Despite their close similarity to normal haematopoietic stem cells, there is increasing evidence that several surface antigens seem to be preferentially expressed in LSCs. For example, the adhesion molecules VLA-4, CLL1, CD44, CD47, CD96 and CD123 (IL-3Rα) could also be considered potential therapeutic targets [40]. Using leukaemia mouse models, several groups have provided proof of principle that targeted interference with monoclonal antibodies against such LSC-surface antigens as CD44, CD123 or CD47 is able to reduce the burden of leukaemia-initiating cells [40, 41].
Epigenetic factors in leukaemogenesis
In addition to structural genetic lesions, epigenetic changes may play an important role in the development and/or maintenance of leukaemia. Epigenetic processes include potentially reversible biochemical modifications of the chromatin either to the DNA or to its associated protein complexes (such as histones) that affect gene expression without alteration of the primary DNA sequence. Leukaemic fusion proteins derive from chromosomal translocations and can also mediate epigenetic silencing or activation of gene expression. In addition, aberrant epigenetic DNA methylation or modification of chromatin-associated histones at specific sites may functionally cooperate in leukaemogenesis. Distinct histone modification patterns have recently been described in murine and human leukaemic cells carrying MLL-fusion oncogenes [42]. New technologies allowing a genome-wide comparison of target genes and associated enzymatic (and potentially druggable) histone modifications may lead to the identification of new therapeutic targets [43]. Aberrant DNA methylation seems to be a dominant mechanism in the progression from myelodysplasia (MDS) to AML [44]. Several epigenetically active compounds such as histone deacetylase or DNA methyltransferase inhibitors are currently being evaluated in several therapeutic trials for MDS, AML and other cancers [45]. Despite their rather nonspecific nature, these agents appear to have a therapeutic potential especially when applied in combination with conventional chemotherapy [45].
Histone modifications generally control the cellular transcriptional activity, and they also regulate micro RNAs (miRNAs). miRNAs are small (18–22 nucleotides) RNA molecules that can negatively control a large number of target genes via a post-transcriptional mechanism. There are currently over 700 human miRNAs deposited in the miRNA database from the Sanger Genome Center (http://www.mirbase.org). Recent data has proposed that aberrant miRNA expression may play a vital role in the pathology of acute leukaemia [46, 47]. Future high-throughput analysis of the entire "miRNAome” will most probably not only improve our understanding of the pathogenesis of the disease, but also furnish new prognostic biomarkers. Through the development of new delivery strategies such as engineered nanoparticles it is likely that miRNA-based technologies will also lead to new therapeutic applications.
New screens – new potential targets – outlook
Powerful molecular screening tools will soon allow us to identify all genetic and epigenetic aberrations that are present in a leukaemic blast. There will be a large number of new alterations in genes and epigenetic regulators in addition to the most prevalent known alterations. The significant heterogeneity of the human genome will force us to analyse a large number of patients, to identify the most prevalent lesions. New cellular and animal models are urgently necessary to functionally validate which of these alterations are essential or negligible for the induction and/or maintenance of a leukaemic phenotype and, eventually, which can be used as potential therapeutic targets. These models will be used for extensive chemical and genetic screens that are able to identify a large number of novel therapeutic targets. Libraries containing several short interfering RNAs (RNAi) able to knock down the expression of all, mouse, fly or human genes, will facilitate the identification of potential targets. Tyner and coworkers have recently developed an RNAi-assisted protein identification (RAPID) platform to search for novel targets to treat leukaemia in patients [48]. Focusing on protein tyrosine kinases (PTKs), they found patient-specific sensitivity by knockdown of PTKs such as FLT1, CSFR1, PDGFR, ROR1, and others [48]. An alternative to identifying oncogenes as direct therapeutic targets is to screen for genes that are essential ("synthetically lethal”) only in the presence of a specific cancer-causing mutation. High-throughput RNAi-based screen for synthetic lethal interactions in cells harbouring a mutant KRAS resulted in the identification of several potential druggable targets, including protein kinases such as STK33 or TBK1 [49, 50]. These studies demonstrated that RNAi screening for functional dependencies resulting from oncogenic mutations might allow identification of new therapeutic targets in tumours with genetic lesions that are up-to-date "undruggable”. Following the paradigm of synthetic lethality, small molecule inhibitors targeting STK33 or TBK1 would be a promising therapeutic approach for leukaemia (and other cancers) with activating RAS mutations (fig. 4).
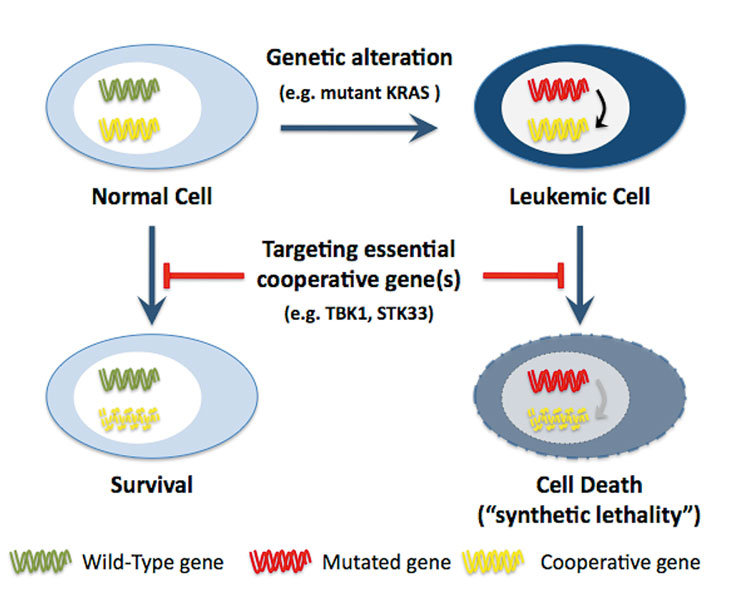
Figure 4
The concept of synthetic lethality in acute leukaemia.
Targeting gene(s) that are functionally linked to the mutated gene should allow the induction of cell death in leukaemic cells but not in normal cells where this gene is not essential.
Despite these very promising advances in our understanding of the biology of the disease, there are still a few drawbacks to be considered. Functional studies using cellular and animal leukaemia models will have to validate the candidates resulting from genome-wide analysis, in order to verify which mutation is indeed necessary for the induction and/or maintenance of the disease. Closer interaction between academic research laboratories with the leading biopharmaceutical companies will be essential to promote the use of promising compounds through clinical trials, and to determine which patients can profit most. Although the complex biology of acute leukaemia makes it rather unlikely that the success of tyrosine kinase inhibitors for CML will easily be repeated, there is hope that our research will provide new clues for targeted therapeutics that will improve acute leukaemia patients’ prognosis and quality of life.
We would like to apologise to our colleagues for failing to cite their important contributions due to strict space constraints. J.S. would like to thank all former and current members of the laboratory for their efforts.
Correspondence:
Juerg Schwaller MD
University Hospital Basel
Department of Biomedicine
ZLF, Lab 318
Hebelstrasse 20
4031 Basel
Switzerland
J.Schwaller@unibas.ch
References
1 Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med. 2006;354(2):166–78.
2 Shipley JL, Butera JN. Acute myelogenous leukemia. Exp Hematol. 2009;37(6):649–58.
3 Chalandon Y, Schwaller J. Targeting mutated protein tyrosine kinases and their signaling pathways in hematologic malignancies. Haematologica. 2005;90(7):949–68.
4 Scholl C, Gilliland DG, Frohling S. Deregulation of signaling pathways in acute myeloid leukemia. Semin Oncol. 2008;35(4):336–45.
5 De Keersmaecker K, Marynen P, Cools J. Genetic insights in the pathogenesis of T-cell acute lymphoblastic leukemia. Haematologica. 2005;90(8):1116–27.
6 Aifantis I, Raetz E, Buonamici S. Molecular pathogenesis of T-cell leukaemia and lymphoma. Nat Rev Immunol. 2008;8(5):380–90.
7 Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer. 2007;7(11):823–33.
8 Moore MA, Chung KY, Plasilova M, Schuringa JJ, Shieh JH, Zhou P, et al. NUP98 dysregulation in myeloid leukemogenesis. Ann N Y Acad Sci. 2007;1106:114–42.
9 Armstrong SA, Kung AL, Mabon ME, Silverman LB, Stam RW, Den Boer ML, et al. Inhibition of FLT3 in MLL. Validation of a therapeutic target identified by gene expression based classification. Cancer Cell. 2003;3(2):173–83.
10 Mullighan CG, Downing JR. Genome-wide profiling of genetic alterations in acute lymphoblastic leukemia: recent insights and future directions. Leukemia. 2009;23(7):1209–18.
11 Walter MJ, Payton JE, Ries RE, Shannon WD, Deshmukh H, Zhao Y, et al. Acquired copy number alterations in adult acute myeloid leukemia genomes. Proc Natl Acad Sci USA. 2009;106(31):12950–5.
12 Papaemmanuil E, Hosking FJ, Vijayakrishnan J, Price A, Olver B, Sheridan E, et al. Loci on 7p12.2, 10q21.2 and 14q11.2 are associated with risk of childhood acute lymphoblastic leukemia. Nat Genet. 2009;41(9):1006–10.
13 Trevino LR, Yang W, French D, Hunger SP, Carroll WL, Devidas M, et al. Germline genomic variants associated with childhood acute lymphoblastic leukemia. Nat Genet. 2009;41(9):1001–5.
14 Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, et al. Recurring Mutations Found by Sequencing an Acute Myeloid Leukemia Genome. N Engl J Med. 2009.
15 Knapper S. FLT3 inhibition in acute myeloid leukaemia. Br J Haematol. 2007;138(6):687–99.
16 Palomero T, Sulis ML, Cortina M, Real PJ, Barnes K, Ciofani M, et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat Med. 2007;13(10):1203–10.
17 Real PJ, Tosello V, Palomero T, Castillo M, Hernando E, de Stanchina E, et al. Gamma-secretase inhibitors reverse glucocorticoid resistance in T cell acute lymphoblastic leukemia. Nat Med. 2009;15(1):50–8.
18 Moellering RE, Cornejo M, Davis TN, Del Bianco C, Aster JC, Blacklow SC, et al. Direct inhibition of the NOTCH transcription factor complex. Nature. 2009;462(7270):182–8.
19 Benekli M, Baumann H, Wetzler M. Targeting Signal Transducer and Activator of Transcription Signaling Pathway in Leukemias. J Clin Oncol. 2009.
20 Schwaller J, Parganas E, Wang D, Cain D, Aster JC, Williams IR, et al. Stat5 is essential for the myelo- and lymphoproliferative disease induced by TEL/JAK2. Mol Cell. 2000;6(3):693–704.
21 Mikkers H, Allen J, Knipscheer P, Romeijn L, Hart A, Vink E, et al. High-throughput retroviral tagging to identify components of specific signaling pathways in cancer. Nat Genet. 2002;32(1):153–9.
22 Amaravadi R, Thompson CB. The survival kinases Akt and Pim as potential pharmacological targets. J Clin Invest. 2005;115(10):2618–24.
23 Shah N, Pang B, Yeoh KG, Thorn S, Chen CS, Lilly MB, et al. Potential roles for the PIM1 kinase in human cancer – a molecular and therapeutic appraisal. Eur J Cancer. 2008;44(15):2144–51.
24 Adam M, Pogacic V, Bendit M, Chappuis R, Nawijn MC, Duyster J, et al. Targeting PIM kinases impairs survival of hematopoietic cells transformed by kinase inhibitor-sensitive and kinase inhibitor-resistant forms of Fms-like tyrosine kinase 3 and BCR/ABL. Cancer Res. 2006;66(7):3828–35.
25 Hu YL, Passegue E, Fong S, Largman C, Lawrence HJ. Evidence that the Pim1 kinase gene is a direct target of HOXA9. Blood. 2007;109(11):4732–8.
26 Pogacic V, Bullock AN, Fedorov O, Filippakopoulos P, Gasser C, Biondi A, et al. Structural analysis identifies imidazo[1,2-b]pyridazines as PIM kinase inhibitors with in vitro antileukemic activity. Cancer Res. 2007;67(14):6916–24.
27 Fedorov O, Marsden B, Pogacic V, Rellos P, Muller S, Bullock AN, et al. A systematic interaction map of validated kinase inhibitors with Ser/Thr kinases. Proc Natl Acad Sci USA. 2007;104(51):20523–8.
28 Chen LS, Redkar S, Bearss D, Wierda WG, Gandhi V. Pim kinase inhibitor, SGI-1776, induces apoptosis in chronic lymphocytic leukemia cells. Blood. 2009;114(19):4150–7.
29 Schwaller J, Frantsve J, Aster J, Williams IR, Tomasson MH, Ross TS, et al. Transformation of hematopoietic cell lines to growth-factor independence and induction of a fatal myelo- and lymphoproliferative disease in mice by retrovirally transduced TEL/JAK2 fusion genes. EMBO J. 1998;17(18):5321–33.
30 Grundler R, Brault L, Gasser C, Bullock AN, Dechow T, Woetzel S, et al. Dissection of PIM serine/threonine kinases in FLT3-ITD-induced leukemogenesis reveals PIM1 as regulator of CXCL12-CXCR4-mediated homing and migration. J Exp Med. 2009;206(9):1957–70.
31 Nilsson SK, Simmons PJ, Bertoncello I. Hemopoietic stem cell engraftment. Exp Hematol. 2006;34(2):123–9.
32 Spiegel A, Kalinkovich A, Shivtiel S, Kollet O, Lapidot T. Stem cell regulation via dynamic interactions of the nervous and immune systems with the microenvironment. Cell Stem Cell. 2008;3(5):484–92.
33 Burger JA, Kipps TJ. CXCR4: a key receptor in the crosstalk between tumor cells and their microenvironment. Blood. 2006;107(5):1761–7.
34 Burger JA, Peled A. CXCR4 antagonists: targeting the microenvironment in leukemia and other cancers. Leukemia. 2009;23(1):43–52.
35 Nervi B, Ramirez P, Rettig MP, Uy GL, Holt MS, Ritchey JK, et al. Chemosensitization of acute myeloid leukemia (AML) following mobilization by the CXCR4 antagonist AMD3100. Blood. 2009;113(24):6206–14.
36 Buonamici S, Trimarchi T, Ruocco MG, Reavie L, Cathelin S, Mar BG, et al. CCR7 signalling as an essential regulator of CNS infiltration in T-cell leukaemia. Nature. 2009;459(7249):1000–4.
37 Kiel MJ, Morrison SJ. Uncertainty in the niches that maintain haematopoietic stem cells. Nat Rev Immunol. 2008;8(4):290–301.
38 Rizo A, Vellenga E, de Haan G, Schuringa JJ. Signaling pathways in self-renewing hematopoietic and leukemic stem cells: do all stem cells need a niche? Hum Mol Genet. 2006;15 Spec No 2:R210–9.
39 Wang JC, Dick JE. Cancer stem cells: lessons from leukemia. Trends Cell Biol. 2005;15(9):494–501.
40 Chan WI, Huntly BJ. Leukemia stem cells in acute myeloid leukemia. Semin Oncol. 2008;35(4):326–35.
41 Lane SW, Scadden DT, Gilliland DG. The leukemic stem cell niche: current concepts and therapeutic opportunities. Blood. 2009;114(6):1150–7.
42 Slany RK. The molecular biology of mixed lineage leukemia. Haematologica. 2009;94(7):984–93.
43 Neff T, Armstrong SA. Chromatin maps, histone modifications and leukemia. Leukemia. 2009;23(7):1243–51.
44 Jiang Y, Dunbar A, Gondek LP, Mohan S, Rataul M, O’Keefe C, et al. Aberrant DNA methylation is a dominant mechanism in MDS progression to AML. Blood. 2009;113(6):1315–25.
45 Bhalla KN. Epigenetic and chromatin modifiers as targeted therapy of hematologic malignancies. J Clin Oncol. 2005;23(17):3971–93.
46 Yendamuri S, Calin GA. The role of microRNA in human leukemia: a review. Leukemia. 2009;23(7):1257–63.
47 Marcucci G, Radmacher MD, Maharry K, Mrozek K, Ruppert AS, Paschka P, et al. MicroRNA expression in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008;358(18):1919–28.
48 Tyner JW, Deininger MW, Loriaux MM, Chang BH, Gotlib JR, Willis SG, et al. RNAi screen for rapid therapeutic target identification in leukemia patients. Proc Natl Acad Sci USA. 2009;106(21):8695–700.
49 Scholl C, Frohling S, Dunn IF, Schinzel AC, Barbie DA, Kim SY, et al. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell. 2009;137(5):821–34.
50 Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009.