Airway smooth muscle cells respond directly to inhaled environmental factors
DOI: https://doi.org/10.4414/smw.2010.13066
Michael
Roth, Michael
Tamm
Pneumology and Pulmonary Cell Research, University Hospital Basel, Basel, Switzerland
Summary
A misled or overreacting immune response is assumed to be the major cause of the most prevalent chronic inflammatory lung diseases, asthma and chronic obstructive pulmonary disease (COPD). The contribution of tissue forming cells, especially of airway smooth muscle cells, to the pathologies of both diseases has only recently attracted some attention. New studies in childhood asthma and a rhesus monkey model strongly suggest a central role of the airway smooth muscle cells in lung development, structure, function and response to environmental factors. Airway smooth muscle cells express and respond to activation of IgE receptors. In addition, airway smooth muscle cells recognise and respond to environmental factors, including allergens and dust, via mechanisms that are independent of the immune system such as PAR2 or calreticulin. Interestingly, these changes occur not on the level of gene activity but on the level of protein synthesis. The reason why these temporary changes become chronic in asthma and COPD remains to be studied.
Introduction
The two most common chronic inflammatory airway diseases are asthma and chronic obstructive pulmonary disease (COPD), which are a substantial burden for healthcare systems worldwide. Together the two diseases affect 11–19% of the world’s population, with wide geographic variation. Asthma and COPD account for the majority of absences from school and work, as well as for increased morbidity and mortality [1, 2].
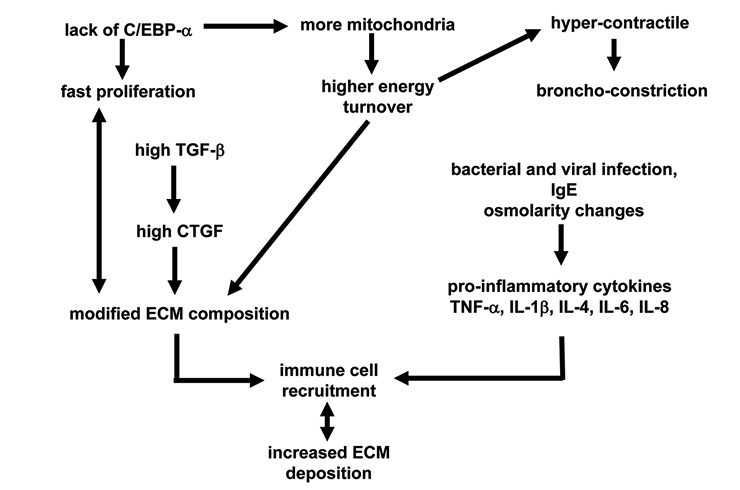
Figure 1
Summary of cell type specific pathologies of isolated human airway smooth muscle cells obtained from patients with asthma when compared to cells isolated from either healthy controls or patients with chronic obstructive pulmonary disease. ECM: extracellular matrix,
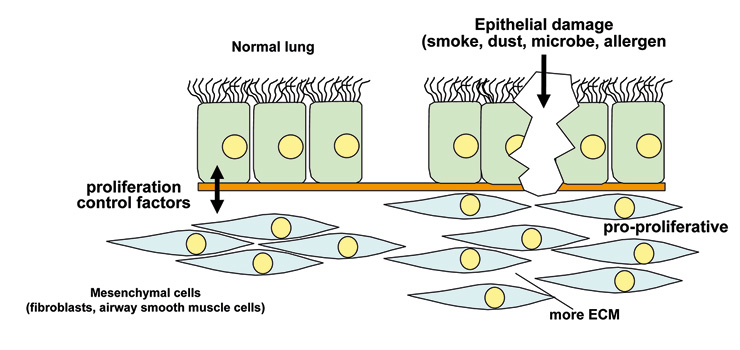
Figure 2
Tissue homoeostatsis is guaranteed by the interaction between epithelial cells and mesenchymal cells. Epithelial damage by mechanical stress or allergens or microorganisms can disrupt this interaction and lead to chronic inflammation.
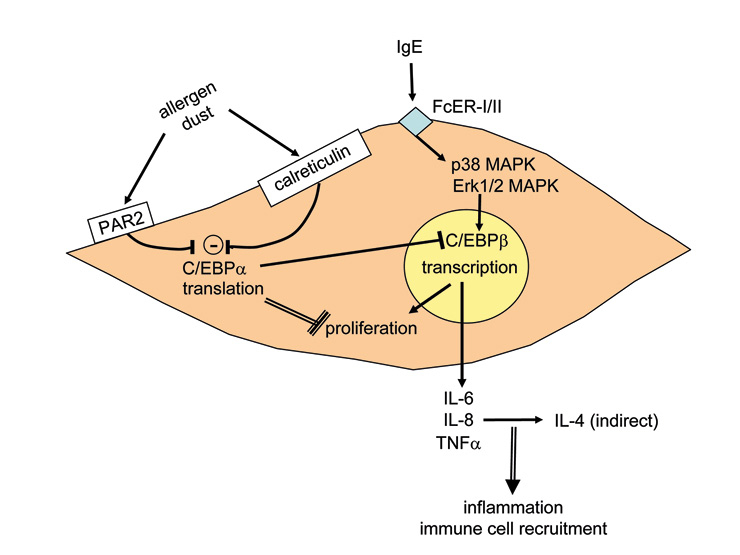
Figure 3
Known mechanisms that modify airway smooth muscle cell action and its implication for the pathogenesis of asthma. FcεR: IgE receptor; MAPK: mitogen-activated protein kinase.
The healthy lung’s main function – gas exchange – relies on homoeostatsis of the tissue structure of the bronchi and of the alveoli, which are formed by very thin cell layers that provide a large surface between inhaled air and blood vessels [3]. Any change of this structure may reduce breathing capacity and thereby gas exchange. Fibrosis of the alveoli with increased extracellular matrix deposition hinders gas exchange. Thickening of the airway wall by tissue-forming cell types including fibroblasts and airway smooth muscle cells, as well as increased deposition of extracellular matrix increases stiffness and hyperconstriction, thereby reducing the breathing capacity [1–3]. While the structure of aveoli is changed in COPD, airway wall remodelling occurs in a disease-specific fashion in the upper airways in asthma and in the smaller airways in COPD [3]. Exacerbation in asthma and COPD is caused by either infections or air pollutants. Air pollution has been estimated to account annually for >3.6m deaths and 62 000 hospital admissions for respiratory malfunction in Europe [4]. Air pollutants impair lung function and cause inflammation and airway remodelling in adults, while in children it may result in reduced lung growth [4].
Recent knowledge of the pathologies of both diseases suggests that a misled immune response is the major cause [2–7]. For years this view has placed the focus of research on lung-infiltrating immune cells and the array of pro-inflammatory cytokine these cells release locally. Data from animal and clinical studies suggested various hypotheses as to how an imbalance of pro-inflammatory cytokines and growth factors induces and propagates chronic inflammatory lung diseases [5–9]. The role of Th1 and Th2 cells was suggested by animal models and seemed to be confirmed in human studies [9–14], resulting in inclusion of this definition in World Health Organisation guidelines [15]. However, subsequent studies did not support this theory since in humans the suspected cytokines (IL-4, IL-5, IL-13) did not correlate with the severity of the diseases and thus put in question the role of T-cells in these pathologies [16–19].
A summary of the large amount of data now available on the role of immune cells in chronic inflammatory lung diseases affords growing evidence that initiation and progression does not necessarily need the immune system [5, 7, 20–22]. At least in childhood asthma, in humans as well as in rhesus monkeys, there is evidence that airway wall remodelling occurs long before any sign of inflammation can be detected [20–22]. Furthermore, the rhesus monkey model suggests a special role for airway smooth muscle in the attraction and activation of lung infiltrating immune cells [20, 22]. This reposes the question of where it all starts?
Asthma was first described as a pathology of the airway smooth muscle since all asthma cases examined presented one unique pathology, an increased mass of airway smooth muscle bundles [23]. Today an increasing number of publications have provided evidence on both the clinical and experimental level that this tissue- and structure-forming cell type may indeed be more important than was subsequently thought. Shortly after it became possible to isolate and cultivate human airway smooth muscle cells, a number of new cell-type and disease-specific pathophysiologies were described. Airway smooth muscle cells of asthma patients show a predisposition to proliferate faster [24, 25] which was linked to both reduced expression of the transcription and differentiation factor C/EBP-α [26] and to an increased number of mitochrondria which were hyperactive [25]. Interestingly, many mitochondrial genes are controlled by C/EBP, which itself is the target of mitochondrial proteins as summarised earlier [27]. In a new mouse model of maternally transmitted airway inflammation it was also shown that the airway smooth muscle cells play a pivotal directing role in lung structural development during embryogenesis, implying this role in adult life during regeneration or inflammation [28]. An overview of these functions is provided in figure 1.
However, asthma is a complex disease with a highly variable phenotype, and it would be superficial to assume that a single cell type may cause it. It is more likely that the communication network linking the different cell types of the airway wall has gone out of balance. However, the airway smooth muscle cell and the so-called myo-fibroblast may be more in the centre of the action than previously assumed [29–31]. New evidence suggests that in asthma epithelial cell differentiation may be less prominent and result in a predisposition to undergo transition to mesenchymal cells, which in turn present features of muscle-like cells [32]. Together with the altered airway smooth muscle cells and myo-fibroblasts, reduced airway epithelial cell function and the lung infiltrated immune cells may keep the chronically inflamed lung in a constant state of alert, thus responding faster to environmental stimuli [20, 22].
In COPD airway remodelling occurs more distally in the medium- to small-size airways, and the cytokines involved are not all the same as in asthma [33–36]. COPD was long regarded as an extreme form of asthma, followed by lung deterioration and emphysema [37, 38]. Again, immune cells were seen as the major driving force behind disease progression. Similarly to asthma, recent clinical observations and experimental data suggest a different story: evidence is accumulating that the interaction of the so-called “epithelial-mesenchymal unit” is disturbed in COPD [20, 37, 38]. The control of an organ’s homoeostasis and function depends on the communication between the different cell types forming it. In this case it is communication between the epithelial cells and the fibroblasts and myo-fibroblasts (fig. 2) in particular. It is now suspected that this communication is disturbed, mainly by products contained in cigarette smoke and by fine dust particles [39, 40]. The question is: How can inhaled substances so different from each other modify the function of specific lung cells persistently?
There is evidence that the airway smooth muscle initiates even the inflammatory response in asthma, especially in response to inhaled allergens and infectious microorganisms [41, 42]. However, on this view it is assumed that inhaled allergens mainly act on immune cells, causing them to adhere to airway smooth muscle cells and thereby altering their function via cell adhesion molecules (CAM). Importantly, it was also pointed out that airway smooth muscle cells release chemo-attractants and thus contribute to the further recruitment of immune cells into the lung [41, 42]. Thus, activated immune cells modify the function of airway smooth muscle cells in such a way that they call in more immune cells.
Microorganisms such as Chlamydia or Rhino-virus are a major cause of exacerbation in asthma and COPD, and they may activate airway smooth muscle cells directly via toll-like receptors (TLR) and glycol proteins. TLR recognise bacterial and viral proteins and stimulate an inflammatory response. For airway smooth muscle cells it appears that LPS is the most important direct ligand to TLR and thus increases airway hyper-responsiveness [43] and the secretion of pro-inflammatory cytokines such as IL-6, eotaxin, or ICAM [44–46]. The observation that microorganisms modulate the activity of differentiation and inflammation regulating transcription factors such as C/EBPs, NFκB, AP-1, and even the glucocorticoid receptor in various host cell types [47–49], requires investigation of this issue in airway smooth muscle cells in asthma and COPD. Such a mechanism could be regarded as a microorganism-induced reprogramming of the host cells’ function and thereby induce a state of chronic inflammation.
Several studies have demonstrated that under defined conditions airway smooth muscle cells of asthma patients produce more pro-inflammatory cytokines than cells of non-asthmatics [50–53]. The range of pro-inflammatory factors released by asthma patients’ activated airway smooth muscle cells includes those which attract immune cells into the lung [35, 41, 42, 54–56] and a differently composed extracellular matrix [50–53]. Changes of the extracellular matrix also have significant effects on the differentiation and function of neighbouring cells and of lung-infiltrating immune cells [53>, 54], although more studies in this area are needed.
These findings point towards a precondition of the airway smooth muscle cell in asthma that has to be activated by environmental factors such as allergens and dust particles, leading to chronic inflammation. The inheritable component of asthma may be linked to innate immunity, since in several studies the expression and function of anaphylatoxin receptor C3a correlated with asthma symptoms in the lungs of mice and humans [57–59]. The C3a receptor is thought to mediate the interaction of airway smooth muscle cells with mast cells and induce degranulation, which contributes to inflammation [57, 59]. However, the conclusion on the role of C3a and C5a in airway smooth muscle cell function in asthma was based on indirect evidence [56] and a later study questioned whether human airway smooth muscle cells express this protein while they interact with mast cells that express C3a and C5a [57].
Studies in a non-human primate model for asthma indicated that exposure to allergens shortly after birth somehow persistently modifies the differentiation setting of airway smooth muscle cells [20, 22, 60]. The studies suggested that the airway smooth muscle cell is able to directly recognise and respond to allergens. Indeed, there are reports that airway smooth muscle cells express the IgE receptors. The FcepsilonRII (CD23) is expressed at a significantly higher level on airway smooth muscle cells of asthma patients compared to controls [61]. In our own study we found no significant differences of the IgE receptor expression comparing them on airway smooth muscle cells of asthma patients and non-asthma controls [62]. In response to IgE complex stimulation, airway smooth muscle cells produced IL-1β, a well known pro-inflammatory factor for asthma. In addition, human airway smooth muscle cells express FcepsilonRI and its activation increases intracellular calcium levels and cell contractility. FcepsilonRI activation also resulted in the production of typical asthma related cytokines including IL-4, IL-5, IL-13 and eotaxin. All these responses were counteracted by the presence of neutralising antibodies to FcepsilonRI [63]. Further, we observed that both cell types secreted asthma-relevant IL-4, IL-6, IL-8 and TNF-α upon IgE stimulation, which was inhibited by IgE-antibodies [62]. Thus, it is likely that earlier reports that sensitisation of airway smooth muscle cells by serum led to increased calcium and muscle cell contraction involved the action of IgE receptors [64, 65]. Also, airway smooth muscle cells expressed the three receptors for IgG (CD64, CD32, CD16), but with no known consequences [61, 66].
It is also possible that airway smooth muscle cells are activated by inhaled allergens or microorganisms by immune-globulin receptor-independent mechanisms which apply to both asthma and COPD. Chambers et al. reported that human airway smooth muscle cells express the protease-activated receptor-2 (PAR-2) and its activation caused muscle cell constriction [67]. This study also suggested an interaction of the IgE receptor pathway with PAR-2, since trypsin increased calcium release and proliferation in serum sensitised cells more than in non-sensitised cells. The same group reported later that PAR-2 activation in human airway smooth muscle cells led to increased secretion of PGE-2 and cyclo-oxygenase-2 [68]. Furthermore, Inflammation up-regulated the expression of PAR-2 by airway smooth muscle cells [69]. Importantly, most PAR-2 mediated responses of airway smooth muscle cells were not inhibited by glucocorticoids [70]. House dust mite allergens activated PAR-2 in epithelial cells [69], modulated the function of chloride channels [71] and stimulated the secretion of IL-6 and IL-8 [73, 74]. Unfortunately, animal models of PAR-2 function in airway inflammation contradict each other and no conclusions can be drawn. A summary of the function and possible interactions of PAR-2, calreticulin and IgE receptors on the function of airway smooth muscle cells is provided in figure 3.
Importantly, house dust mite allergens disrupted the cell-cell contacts and created gaps in the epithelial cell barrier; this process involved the action of PAR-2 and may enable allergens to penetrate into sub-epithelial tissue [75]. How this pro-cess is linked to other allergen transport mechanisms across the epithelial barrier is unclear. However, there is evidence that certain splice-forms of the IgE receptor FcεRI, which is expressed by epithelial cells, bind and transport allergens into the sub-epithelial tissue [76]. There is also evidence that at least FcεRI is a binding partner for allergens and transports them through epithelial cells in large quantities without changing their structure [77]. By such mechanisms allergens can cross the epithelial barrier and act in sub-epithelial tissues.
In summary, these data strongly suggest that airway smooth muscle cells take an active part in the immune response, far from being only a responder to activated immune cells. Airway smooth muscle cells contribute to chronic airway inflammation by secreting pro-inflammatory, chemo-attractive cytokines, and respond to allergens via immune globulin receptors and non-immune systems. Thus they modify the lung’s response to environmental factors leading to chronic inflammation. However, the main questions (how and why such short term changes of physiology in normal cells become persistent in cells of asthma and COPD patients) remain to be investigated.
Michael Roth MD
Lab 305, Pulmonary Cell Research
Dept.
Biomedicine
University Hospital Basel
Petersgraben 4
CH-4031 Basel
rothmic[at]uhbs.ch
References
1 Eder W, Ege MJ, von Mutius E. The asthma epidemic. N Engl J Med. 2006;355:2226–35.
2 Fabbri L, Peters SP, Pavord I, Wenzel SE, Lazarus SC, Macnee W, et al. Allergic rhinitis, asthma, airway biology, and chronic obstructive pulmonary disease in AJRCCM in 2004. Am J Respir Crit Care Med. 2005;171:686–98.
3 Weibel ER. What makes a good lung? Swiss Med Wkly. 2009;139:375–86.
4 Künzli N, Perez L. Evidence based public health – the example of air pollution. Swiss Med Wkly. 2009;139:242–50.
5 Holgate ST. Pathogenesis of asthma. Clin Exp Allergy. 2008;38:872–97.
6 Greenberger PA. 7. Immunologic lung disease. J Allergy Clin Immunol. 2008;121(2 Suppl):S393–7.
7 London SJ. Gene-air pollution interactions in asthma. Proc Am Thorac Soc. 2007;4:217–20.
8 Cohn L, Homer RJ, Niu N, Bottomly K. T helper 1 cells and interferon gamma regulate allergic airway inflammation and mucus production. J Exp Med. 1999;190:1309–18.
9 Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. 1986;136:2348–57.
10 Hamid Q, Azzawi M, Ying S, Moqbel R, Wardlaw AJ, Corrigan CJ, et al. Expression of mRNA for interleukin-5 in mucosal bronchial biopsies from asthma. J Clin Invest. 1991;87:1541–6.
11 Robinson DS, Hamid Q, Ying S, Tsicopoulos A, Barkans J, Bentley AM, et al. Predominant TH2-like bronchoalveolar T-lymphocyte population in atopic asthma. N Engl J Med. 1992;326:298–304.
12 Ying S, Durham SR, Corrigan CJ, Hamid Q, Kay AB. Phenotype of cells expressing mRNA for TH2-type (interleukin 4 and interleukin 5) and TH1-type (interleukin 2 and interferon gamma) cytokines in bronchoalveolar lavage and bronchial biopsies from atopic asthmatic and normal control subjects. Am J Respir Cell Mol Biol. 1995;12:477–87.
13 Umetsu DT, DeKruyff RH. Th1 and Th2 CD4+ cells in the pathogenesis of allergic diseases. Proc Soc Exp Biol Med. 1997;215:11–20.
14 Chapman RW. Canine models of asthma and COPD. Pulm Pharmacol Ther. 2008;21:731–42.
15 WHO/NHLBI Workshop Report. Global strategy for asthma management and prevention. Bethesda, MD: National Institute of Health, National Heart, Lung, and Blood Institute; 1995. Publication No. 95–3659.
16 Sano T, Nakamura Y, Matsunaga Y, Takahashi T, Azuma M, Okano Y, et al. FK506 and cyclosporin A inhibit granulocyte/macrophage colony-stimulating factor production by mononuclear cells in asthma. Eur Respir J. 1995;8:1473–8.
17 Borger P, Kauffman HF, Postma DS, Vellenga E. Interleukin-4 gene expression in activated human T lymphocytes is regulated by the cyclic adenosine monophosphate-dependent signaling pathway. Blood. 1996;87:691–8.
18 Sihra BS, Kon OM, Durham SR, Walker S, Barnes NC, Kay AB. Effect of cyclosporin A on the allergen-induced late asthmatic reaction. Thorax. 1997;52:447–52.
19 Leckie MJ, ten Brinke A, Khan J, Diamant Z, O’Connor B, Walls C, et al. Effects of an interleukin-5 blocking monoclonal antibody on eosinophilic airway hyper-responsiveness and the late asthmatic response. Lancet. 2000;356:2144–8.
20 Plopper CG, Hyde DM. The non-human primate as a model for studying COPD and asthma. Pulm Pharmacol Ther. 2008;21:755–66.
21 Jenkins HA, Cool C, Szefler SJ, Covar R, Brugman S, Gelfand EW, et al. Histopathology of severe childhood asthma: a case series. Chest. 2003;124:32–41.
22 Miller LA, Gerriets JE, Tyler NK, Abel K, Schelegle ES, Plopper CG, et al. Ozone and allergen exposure during postnatal development alters the frequency and airway distribution of CD25+ cells in infant rhesus monkeys. Toxicol Appl Pharmacol. 2009;236:39–48.
23 Huber H, Koesser K. The pathology of bronchial asthma. Arch Intern Med. 1922;30:689–760.
24 Johnson PR, Roth M, Tamm M, Hughes M, Ge Q, King G, Burgess JK, Black JL. Airway smooth muscle cell proliferation is increased in asthma. Am J Respir Crit Care Med. 2001;164:474–7.
25 Trian T, Benard G, Begueret H, Rossignol R, Girodet PO, Ghosh D, et al. Bronchial smooth muscle remodeling involves calcium-dependent enhanced mitochondrial biogenesis in asthma. J Exp Med. 2007;204:3173–81.
26 Roth M, Johnson PR, Borger P, Bihl MP, Rüdiger JJ, King GG, et al. Dysfunctional interaction of C/EBPalpha and the glucocorticoid receptor in asthmatic bronchial smooth-muscle cells. N Engl J Med. 2004;351:560–74.
27 Roth M, Black JL. An imbalance in C/EBPs and increased mitochondrial activity in asthmatic airway smooth muscle cells: novel targets in asthma therapy? Br J Pharmacol. 2009;157:334–41.
28 Jesudason EC. Airway smooth muscle: an architect of the lung? Thorax. 2009;64:541–5.
29 Ichikawa T, Sugiura H, Koarai A, Yanagisawa S, Kanda M, Hayata A, et al. Peroxynitrite augments fibroblast-mediated tissue remodeling via myofibroblast differentiation. Am J Physiol Lung Cell Mol Physiol. 2008;295:L800–8.
30 Fernandes DJ, Bonacci JV, Stewart AG. Extracellular matrix, integrins, and mesenchymal cell function in the airways. Curr Drug Targets. 2006;7:567–77.
31 Wicks J, Haitchi HM, Holgate ST, Davies DE, Powell RM. Enhanced upregulation of smooth muscle related transcripts by TGF beta2 in asthmatic (myo) fibroblasts. Thorax. 2006;61:313–9.
32 Hackett TL, Warner SM, Stefanowicz D, Shaheen F, Pechkovsky DV, Murray LA, et al. Induction of Epithelial-Mesenchymal Transition in Primary Airway Epithelial Cells from Asthmatic Patients by TGF{beta}1. Am J Respir Crit Care Med. 2009;180:122–33.
33 Ferdinands JM, Mannino DM. Obstructive lung disease models: what is valid? COPD. 2008;5:382–93.
34 Taylor DR. Risk assessment in asthma and COPD: a potential role for biomarkers? Thorax. 2009;64:261–4.
35 Barnes PJ. The cytokine network in asthma and chronic obstructive pulmonary disease. J Clin Invest. 2008;118:3546–56.
36 Sturton G, Persson C, Barnes PJ. Small airways: an important but neglected target in the treatment of obstructive airway diseases. Trends Pharmacol Sci. 2008;29:340–5.
37 Cosio MG, Saetta M, Agusti A. Immunologic aspects of chronic obstructive pulmonary disease. N Engl J Med. 2009;360:2445–54.
38 Sarir H, Henricks PA, van Houwelingen AH, Nijkamp FP, Folkerts G. Cells, mediators and Toll-like receptors in COPD. Eur J Pharmacol. 2008;585:346–53.
39 Crystal RG, Randell SH, Engelhardt JF, Voynow J, Sunday ME. Airway epithelial cells: current concepts and challenges. Proc Am Thorac Soc. 2008;5:772–7.
40 Thorley AJ, Tetley TD. Pulmonary epithelium, cigarette smoke, and chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2007;2:409–28.
41 Tliba O, Amrani Y, Panettieri RA Jr. Is airway smooth muscle the “missing link” modulating airway inflammation in asthma? Chest. 2008;133:236–42.
42 Tliba O, Panettieri RA Jr. Regulation of inflammation by airway smooth muscle. Curr Allergy Asthma Rep. 2008;8:262–8.
43 Luo SF, Wang CC, Chiu CT, Chien CS, Hsiao LD, Lin CH, et al. Lipopolysaccharide enhances bradykinin-induced signal transduction via activation of Ras/Raf/MEK/MAPK in canine tracheal smooth muscle cells. Br J Pharmacol. 2000;130:1799–808.
44 Sukkar MB, Xie S, Khorasani NM, Kon OM, Stanbridge R, Issa R, et al. Toll-like receptor 2, 3, and 4 expression and function in human airway smooth muscle. J Allergy Clin Immunol. 2006;118:641–8.
45 Morris GE, Whyte MK, Martin GF, Jose PJ, Dower SK, Sabroe I. Agonists of toll-like receptors 2 and 4 activate airway smooth muscle via mononuclear leukocytes. Am J Respir Crit Care Med. 2005;171:814–22.
46 Shan X, Hu A, Veler H, Fatma S, Grunstein JS, Chuang S, et al. Regulation of Toll-like receptor 4-induced proasthmatic changes in airway smooth muscle function by opposing actions of ERK1/2 and p38 MAPK signaling. Am J Physiol Lung Cell Mol Physiol. 2006;291:L324–33.
47 Oliver BG, Lim S, Wark P, Laza-Stanca V, King N, Black JL, et al. Rhinovirus exposure impairs immune responses to bacterial products in human alveolar macrophages. Thorax. 2008;63:519–25.
48 Oliver BG, Johnston SL, Baraket M, Burgess JK, King NJ, Roth M, et al. Increased proinflammatory responses from asthmatic human airway smooth muscle cells in response to rhinovirus infection. Respir Res. 2006;7:71.
49 Gencay MM, Tamm M, Glanville A, Perruchoud AP, Roth M. Chlamydia pneumoniae activates epithelial cell proliferation via NF-kappaB and the glucocorticoid receptor. Infect Immun. 2003;71:5814–22.
50 Klagas I, Goulet S, Karakiulakis G, Zhong J, Baraket M, Black JL, et al. Decreased hyaluronan in airway smooth muscle cells from patients with asthma and COPD. Eur Respir J. 2009;34:616–28.
51 Dekkers BG, Schaafsma D, Nelemans SA, Zaagsma J, Meurs H. Extracellular matrix proteins differentially regulate airway smooth muscle phenotype and function. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1405–13.
52 Slats AM, Janssen K, van Schadewijk A, van der Plas DT, Schot R, van den Aardweg JG et al. Expression of smooth muscle and extracellular matrix proteins in relation to airway function in asthma. J Allergy Clin Immunol. 2008;121:1196–202.
53 Zhang W, Gunst SJ. Interactions of airway smooth muscle cells with their tissue matrix: implications for contraction. Proc Am Thorac Soc. 2008;5:32–9.
54 Johnson PR, Ammit AJ, Carlin SM, Armour CL, Caughey GH, Black JL. Mast cell tryptase potentiates histamine-induced contraction in human sensitized bronchus. Eur Respir J. 1997;10:38–43.
55 Haddon DJ, Antignano F, Hughes MR, Blanchet MR, Zbytnuik L, Krystal G, et al. SHIP1 is a repressor of mast cell hyperplasia, cytokine production, and allergic inflammation in vivo. J Immunol. 2009;183:228–36.
56 Hamid Q, Tulic M. Immunobiology of asthma. Annu Rev Physiol. 2009;71:489–507.
57 Mascia F, Mariani V, Giannetti A, Girolomoni G, Pastore S. House dust mite allergen exerts no direct proinflammatory effects on human keratinocytes. J Allergy Clin Immunol. 2002;109:532–8.
58 Thangam EB, Venkatesha RT, Zaidi AK, Jordan-Sciutto KL, Goncharov DA, Krymskaya VP, et al. Airway smooth muscle cells enhance C3a-induced mast cell degranulation following cell-cell contact. FASEB J. 2005;19:798–800.
59 Humbles AA, Lu B, Nilsson CA, Lilly C, Israel E, Fujiwara Y, et al. A role for the C3a anaphylatoxin receptor in the effector phase of asthma. Nature. 2000;406:998–1001.
60 Plopper CG, Smiley-Jewell SM, Miller LA, Fanucchi MV, Evans MJ, Buckpitt AR et al. Asthma/allergic airways disease: does postnatal exposure to environmental toxicants promote airway pathobiology? Toxicol Pathol. 2007;35:97–110.
61 Hakonarson H, Carter C, Kim C, Grunstein MM. Altered expression and action of the low-affinity IgE receptor FcepsilonRII (CD23) in asthmatic airway smooth muscle. J Allergy Clin Immunol. 1999;104:575–84.
62 Roth M, Tamm M. Omalizumab blocks IgE induced cytokine synthesis by asthmatic airway smooth muscle cells. Annals Allergy, Asthma, Immunol 2009 in press.
63 Gounni AS, Wellemans V, Yang J, Bellesort F, Kassiri K, Gangloff S, et al. Human airway smooth muscle cells express the high affinity receptor for IgE (Fc epsilon RI): a critical role of Fc epsilon RI in human airway smooth muscle cell function. J Immunol. 2005;175:2613–21.
64 Black JL, Marthan R, Armour CL, Johnson PR. Sensitization alters contractile responses and calcium influx in human airway smooth muscle. J Allergy Clin Immunol. 1989;84:440–7.
65 Watson N, Bodtke K, Coleman RA, Dent G, Morton BE, Rühlmann E, et al. Role of IgE in hyperresponsiveness induced by passive sensitization of human airways. Am J Respir Crit Care Med. 1997;155:839–44.
66 Hakonarson H, Grunstein MM. Autologously up-regulated Fc receptor expression and action in airway smooth muscle mediates its altered responsiveness in the atopic asthmatic sensitized state. Proc Natl Acad Sci U S A. 1998;95:5257–62.
67 Chambers LS, Black JL, Poronnik P, Johnson PR. Functional effects of protease-activated receptor-2 stimulation on human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2001;281:L1369–78.
68 Chambers LS, Black JL, Ge Q, Carlin SM, Au WW, Poniris M, et al. PAR-2 activation, PGE2, and COX-2 in human asthmatic and nonasthmatic airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2003;285:L619–27.
69 Freund-Michel V, Frossard N. Inflammatory conditions increase expression of protease-activated receptor-2 by human airway smooth muscle cells in culture. Fundam Clin Pharmacol. 2006;20:351–7.
70 Saleh SM, Mann TS, Peters T, Betts RJ, Henry PJ. Influence of dexamethasone on protease-activated receptor 2-mediated responses in the airways. J Pharmacol Exp Ther. 2008;324:622–30.
71 Asokananthan N, Graham PT, Stewart DJ, Bakker AJ, Eidne KA, Thompson PJ, et al. House dust mite allergens induce proinflammatory cytokines from respiratory epithelial cells: the cysteine protease allergen, Der p 1, activates protease-activated receptor (PAR)-2 and inactivates PAR-1. J Immunol. 2002;169:4572–8.
72 Cho HJ, Choi JY, Yang YM, Hong JH, Kim CH, Gee HY, et al. House dust mite extract activates apical Cl(-) channels through protease-activated receptor 2 in human airway epithelia. J Cell Biochem. 2010;109:1254–63.
73 Adam E, Hansen KK, Astudillo Fernandez O, Coulon L, Bex F, Duhant X, et al. The house dust mite allergen Der p 1, unlike Der p3, stimulates the expression of interleukin-8 in human airway epithelial cells via a proteinase-activated receptor-2-independent mechanism. J Biol Chem. 2006;281:6910–23.
74 Kauffman HF, Tamm M, Timmerman JA, Borger P. House dust mite major allergens Der p 1 and Der p 5 activate human airway-derived epithelial cells by protease-dependent and protease-independent mechanisms. Clin Mol Allergy. 2006;4:5.
75 Heijink IH, van Oosterhout A, Kapus A. EGFR signaling contributes to house dust mite-induced epithelial barrier dysfunction. Eur Respir J. 2010. Epub ahead of print]PMID: 20351035
76 Montagnac G, Yu LC, Bevilacqua C, Heyman M, Conrad DH, Perdue MH, et al. Differential role for CD23 splice forms in apical to basolateral transcytosis of IgE/allergen complexes. Traffic. 2005;6:230–42.
77 Li H, Nowak-Wegrzyn A, Charlop-Powers Z, Shreffler W, Chehade M, Thomas S, et al. Transcytosis of IgE-antigen complexes by CD23a in human intestinal epithelial cells and its role in food allergy. Gastroenterology. 2006;131:47–58.