Figure 1
DOI: https://doi.org/10.4414/smw.2010.13041
A significant amount of medical research in hospitals is conducted on data, especially patient data. For instance, in 2008, at the Geneva University Hospitals (HUG), some 15% of research protocols fell into this category. When the data to be analysed has alreadybeen collected (e.g., by the physician treating the patient), the research is said to be retrospective. Retrospective research can also be conducted on precollected and stored biological samples.
Patients are not necessarily aware that their data or samples will be used for a purpose other than their own medical care. They could be disappointed in the integrity of their physicians if they were to later learn of this research use. Moreover, if patient data is transferred to third parties outside the medical team, the risk that it could be disclosed further, perhaps even outside the medical realm, increases. Adverse consequences are especially likely if sensitive data is disclosed to patients’ employers or insurers. Furthermore, when research is performed on biological material, the sample will be consumed in whole or in part. Should the patient need it for subsequent treatment or diagnostic purposes, the amount remaining may be insufficient. From an ethical perspective, the principle of autonomy commands that individuals be allowed to decide to participate in any kind of research, including health data banks and biobanks [1] and retrospective research (as explained in the international research guidelines of the Council for International Organizations of Medical Sciences [CIOMS]; commentary on guideline 4) [2]. The principle of beneficence dictates that medical treatment be given priority over research objectives [3, 4].
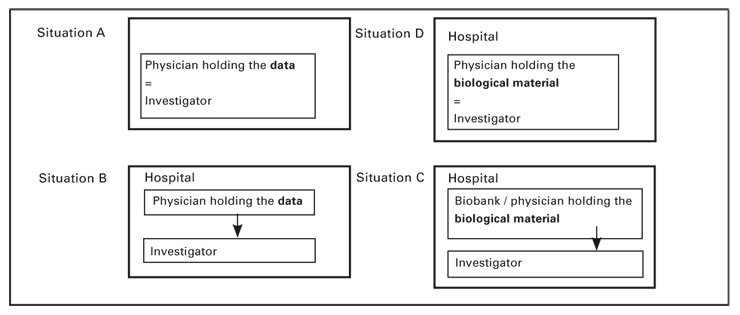
Researchers in Switzerland wishing to access stored data or material need to understand both the law and the ethical considerations before they proceed. They often want to know if they can receive and use data or material without patients’ prior informed consent. Although the answer is far from straightforward, this article seeks to clarify the conditions under which such access is allowed. The first and main chapter focuses on the issue of data and sample access under the four most commonly encountered situations (see fig. 1). The second chapter anticipates the implications of the proposed new federal law on health research (hereafter abbreviated P-LRH from the French title). The final chapter makes suggestions to facilitate retrospective research. Our analysis and recommendations are limited to research done in hospitals by institutional physicians/investigators without a commercial sponsor. More elaborate details on the legal aspects are provided in endnotes to a longer version of this article available on the Web site of Swiss Medical Weekly.
The rules to be observed differ depending on two initial classifications: First, whether it is research data or biological material that is sought and, second, whether the physician holding the data/material is also the investigator. This is why we analyse separately four situations (for an overview, see figure 1 below). In subchapter A, the researcher is also the physician who collected the data from the patient. In subchapter B, the researcher has to obtain the dataset from the physician who treated the patient. In the third and fourth hypotheses, the research implies the use of stored biological material, either transferred from the physician to the researcher (subchapter C) or used by the physician himself (subchapter D). We limit our review to research performed within a hospital, excluding exchanges of data among hospitals. The analysis is based on Swiss laws, hospital-internal regulations and, to a lesser extent, Swiss and international ethical guidelines.
Figure 1
Treating physicians who reopen their own patient files to extract data for research purposes are not breaching medical confidentiality. More accurately, they are not infringing article 321 of the Swiss penal code (CP) [5], which punishes violation of medical confidentiality by up to three years of prison [6, 7].
Even though they are not breaching the CP, they are not free to use the data as they wish. The Swiss federal law on data protection (hereafter abbreviated “LPD” from the French title) [8] sets forth the rules applicable to any kind of data handling, whether by private individuals or by federal authorities (“traitement de données”). However, public cantonal hospitals must abide by the cantonal provisions on data protection, unless the underlying task requires the application of federal law or their cantonal legislation refers back to the LPD.
A cardinal principle of the LPD is that data can only be used in accordance with the purpose initially indicated or implied to the person who provided it. When patients supply data while under medical care, they are entitled to assume that it will be used for treatment purposes only, and not for research purposes. If physicians want to use it for research, the LPD requires, as a general rule, that they contact the patients and obtain their consent (or at least their nonopposition) after informing them of the research contemplated.
The LPD allows for an exception to this rule if the balance of interests favors the data user over the data provider. This is normally the case if patient data is used for research purposes, provided that results are published in a way that does not allow patients’ identification. Physicians must still submit their research projects to the competent ethics committee, because cantonal laws ordinarily require ethics committee to approve all medical research, regardless of its retrospective nature and regardless of the origin of the data (in Geneva, see article 61, paragraph 2, letter f, of the Law on health [9]). Therefore, the protocol must describe the interests at stake and show how the expected benefits of the research will exceed any potential risk or harm to the patient.
The situation is markedly different when the researcher does not already hold the data to be analysed, but needs to access it from physicians. Physicians would breach article 321 CP if they divulged data they collected to the researcher without patients’ consent. The simple act of communicating the patient’s name and address (so as to allow the researcher to initiate the informed consent process) is sufficient to infringe article 321 [10].
There are three main situations where this communication of medical data does not violate the CP: (1) patients have given consent; (2) the data is anonymous, or (3) the special procedure for communication of nonanonymous data for research purposes has been followed.
Patients may allow their physicians to communicate data to third parties, including researchers. The salient question is whether this permission must always meet the high standards related to the informed consent process. In other words, must patients always be informed in detail about the purpose and modalities of the data communication? Or can they simply give blanket consent to any future use of their data for research purposes?
This question has not been affirmatively answered by the courts. With many bioethicists [11–16] and the Swiss Academy of Medical Sciences (SAMS) [17], we are of the opinion that general consent is sometimes sufficient. Patients should at least receive information proportionate to the risks they will be subject to. If these risks are low and/or obvious, a broad consent to future research should be considered satisfactory. Patients always remain free to ask for additional information. They may also refuse to provide general consent, and instead stipulate that only specific projects be allowed to make use of their data.
We would recommend going one step further to suggest that public hospitals systematically submit general consent forms for future research on data whenever a new patient completes the administrative paperwork associated with initiation of a nonurgent medical treatment. Thus, patients’ attention would be drawn to the research activities conducted at public hospitals. From the beginning, patients would be offered an opt-in choice to have their data used for any future research projects. They would be told that research ethics committees would review each of these future projects. If not willing to give blanket consent, they could alternatively signal their willingness to be recontacted later by the researcher, should there be a specific project that necessitates the use of their data.
Article 321 can only be violated if identified or identifiable data is communicated. Therefore, physicians can avoid liability under article 321 CP by transmitting only anonymised data.
In that context, the meaning of “anonymised data” remains equivocal [18]. Anonymous implies that, from the start, data was collected without link to the patient’s identity (e.g., an anonymous survey). Anonymised is used for a dataset stripped of both direct (e.g., name, social security number, sickness insurance number, picture) and indirect patient identifiers (e.g., date of birth, address, profession, physical characteristics). Two kinds of anonymisation are distinguished: irreversible anonymisation where it is impossible or extremely difficult to go back and uncover the identity of the patient; and reversible anonymisation (or coded data) where someone retains the possibility to re-link the data with the corresponding patient through a code. In the second case, the code is typically not made available to the researchers themselves, but kept secure under the control of a third party [19, 20]).
The key question is whether it is enough to reversibly anonymise a dataset to avoid sanctions under article 321 CP. Unfortunately, the courts have not had the opportunity to answer this question. In our view, reversible anonymisation should be sufficient, if the physician has taken all appropriate precautionary measures to protect the code and has pledged not to communicate it to the researchers. In such a situation, researchers will be unable to obtain the code without a security breach; from their point of view, the dataset is completely anonymous.
Anonymisation presents certain drawbacks that explain why many retrospective research projects do not follow this route.
First, anonymisation entails significant work on the part of physicians holding data [10]. They must remove from their files all information items that directly or indirectly identify patients [21]. In view of the workload of university physicians, they will rarely agree to perform this task for the benefit of researchers. Hospitals should consider investing resources to assist physicians who are willing to undertake this task.
Second, certain research projects just cannot be conducted on (irreversibly) anonymised data-sets. This can be the case when individual information found in distinct databases needs to be combined or if future health data needs to be added.
Third, irreversible anonymisation may be problematic for health and ethical reasons, as it deprives the patient of the chance of ultimately receiving (possibly important) research results [17, 19]. This is why the proposed new law on research (P-LRH, at articles 14 and 34) forbids anonymisation if the expected results could help diagnose, treat or prevent serious diseases from which the patient either suffers now or may suffer in the future.
Finally, if patients have given specific instructions forbidding any use of their data, it is not possible to evade them by anonymisation. Although article 321 CP will not be infringed, physicians breaching this commitment made to their patients would nonetheless be liable for their disclosure.
Swiss law permits research on nonanonymised data without patients’ consent under the following exception. Both physicians transferring the data and researchers using it will not violate the law if the conditions below (found in article 321bis CP and in the Ordinance regarding authorisations to lift professional secrecy for medical research (OALSP) [6]), are met.
a) The hospital holds a general authorisation delivered by the Commission of experts on professional secrecy related to medical research (hereafter the Expert Commission).
b) The hospital’s research ethics committee approved the protocol describing the project proposed by the researcher [22].
c) The interest of the planned research exceeds patients’ interest in maintaining the (full) confidentiality of the data.
d) It would be impossible or very difficult to obtain patients’ consent.
e) Patients were informed of the possibility of research being performed on their data and did not object.
f) The researcher undertakes to maintain the personal data as strictly confidential and to use anonymised data to the extent feasible.
Each of these conditions deserves brief explanations.
a) All university hospitals have requested and obtained from the Expert Commission a general authorisation. This gives them general permission to conduct retrospective research within the hospital, provided that the other conditions (b to f) are met. More precisely, the authorisation allows physicians holding information to provide it (if they so wish) to the authorisation beneficiaries, without breaching their civil law and criminal law duty to abide by medical secrecy. Authorisation decisions contain provisions specific to the hospital, but they remain by and large similar. To allow for at least minimal supervision by the Expert Commission, the hospital must make yearly reports announcing all its retrospective research projects that have relied on the general authorisation. The reports are only sent to the Expert Commission and not made public.
b) As for any research project, hospital researchers are required to submit a protocol and all accompanying documents to their ethics committees. Committees should examine research proposals as they would any other protocols (e.g., whether the research is correctly described and scientifically valid). Additionally, committees will decide whether informed consent can be waived; in other words, they will verify whether conditions (c) to (f) are fulfilled.
c) As for any other research endeavor, the expected benefits of the project should exceed the possible risks and inconveniences placed on the subject. When the research is retrospective, the main risks relate to breach of trust and breach of confidentiality. However, referring to the published practice of the Expert Commission, it appears that the bar is not set particularly high: Interesting projects have no difficulties meeting this condition.
d) A waiver of consent is only admissible if it is impossible or very difficult to contact each patient and obtain his or her specific consent for the retrospective research. The reports published by the Expert Commission shed light on how this crucial condition is understood [10, 21]. Valid and invalid grounds are summarised in table 1.
If some patients can easily be contacted, while contacting others would be difficult, it might be justified to require that consent be sought from the first group, while the general authorisation will cover patients from the second group. Moreover, when treating physicians can already anticipate that medical data will be used for research later on, they should inform their patients without delay and obtain their consent; in other words, they should not “game the system” by opting for retrospective research under a consent waiver, instead of prospective research based on full consent.
e) Unless the data was collected before 1996, patients must have been told, typically while still under treatment, of the possibility of research being performed on their data. They must have been advised that they may opt out of this research (“veto right”). Any opt-out must be noted, usually in the patients’ medical file; such opt-out is absolutely binding for the researcher.
The Expert Commission has confirmed that this information is not equivalent to that given for informed consent purposes. Informing potential research subjects recruited to participate in interventional studies is part of an elaborate process, based on both oral and written explanations. On the contrary, information precedent to retrospective research usually consists of a short message in a brochure handed out to the patient. The Expert Commission even allows hospitals to meet the retrospective research requirement by a statement on their Web sites. For example, the Geneva University Hospitals (HUG) indicate on their Web page, under patients’ rights, that:
“The HUG are a university institution where research represents an important activity. It is thanks to research that medical knowledge progresses. You can be invited to participate in a research program or to teaching work; you will be then thoroughly informed and your written consent will be required. You have the right to refuse or to withdraw your consent at any time. But for opposition on your side and in compliance with the law, data collected during your stay may be used for research purposes. These research projects are conducted under conditions guaranteeing strict anonymity and total confidentiality.” (free translation of a text which is available only in French).
This information does not need to be given orally; no discussion between patients and physicians has to take place. Although the HUG internal guidelines state that treating physicians should also inform their patients, in practice this is far from systematic. If patients wish to exercise their opt-out right, they must take the initiative of contacting their medical team or the hospital officials, since there is no online declaration mechanism.
f) As already mentioned, conducting research on anonymous data presents the advantage of bypassing the requirement of informed consent. Yet, if this type of data is not available to the researchers, they may use nonanonymous data. In this case, they are obliged to keep the nonanonymised dataset strictly confidential. Furthermore, they must remove the identifiers as soon as feasible, so as to pursue their work on an (reversibly or irreversibly) anonymised dataset.
The research protocol must describe how confidentiality will be protected and how anonymisation will be achieved. It is the task of ethics committees to control the presence and pertinence of these explanations. It goes without saying that publications resulting from the research should not reveal patients’ identity.
For all retrospective research conducted under the hospital’s general authorisation, researchers and members of their team must sign a standard form reminding them of their confidentiality obligations. The signed pledge is included in the application file submitted to the ethics committee.
| Table 1 |
| – patients are likely to be deceased (except when the death just occurred at the hospital);– patients have probably left Switzerland or there is reason to believe that their current addresses in Switzerland cannot be found;– the number of patients whose data needs to be analysed is very high (e.g., 500);– similarly, patients come from a large geographical area, perhaps because the retrospective research is conducted in several different hospitals;– patients suffer from very serious illnesses, especially psychiatric illnesses, and contacting them would cause additional burden;– similarly, mere contact carries the risk of making other people aware of their conditions (stigmatisation);– patients have died in dramatic circumstances (e.g., death of a premature baby) and asking their legal representatives for consent would be inconsiderate;– to maintain the scientific validity of the research when the size of the population is small, all patient files need to be included in the research;– similarly, to avoid responding bias, it is imperative that data from both patients who consent to the research and patients who object be included. |
| – the patient is held to be incompetent;– the research only represents a change of medical practice (e.g., new way to perform surgery);– the research is too complex to explain to patients (e.g., surgery protocol);– the interest of the research is so high that, for this reason alone, waiver should be granted;– patients would probably refuse their consent;– patients do not like to be reminded of their disease (except if the disease is life-threatening or of a psychiatric nature);– patients do not like to be reminded of their past illegal behavior. |
The explanations above referred to data, i.e. paper or electronic files, whereas biological material is not generally understood to be data. Yet the question arises of whether researchers can – without asking for patients’ prior informed consent – access biological samples already collected and stored within their hospital. To answer the question, we must determine the scope of application of article 321 CP, article 321bis CP, the OALSP and the LPD.
Article 321 CP encompasses anything physicians have learned or accessed in the course of their interactions with their patients. Therefore, biological material that is linked to patients’ identity is without doubt covered by medical secrecy. Physicians who collect such material must maintain it confidential; they can use it for their own research, but should respect the rules of the LPD (see subchapter A above). Even though the LPD also refers to “data”, it has been interpreted broadly to encompass all kind of information.
If the biological material has been made anonymous, then article 321 is simply inapplicable. Not only can physicians use it for their own research, but they can also transfer it to third parties, subject to residual constraints stated in cantonal laws (typically requiring ethics committee’s approval) and in the SAMS directive on biobanks [17]. Although SAMS directives are not legally binding, the Conduct Code of the Swiss Medical Association (FMH) (article 18) [24] has declared them compulsory for Swiss physicians. Violation of the biobank directive could therefore lead to (private) sanctions, but would not necessarily make physicians liable towards their patients. If the material is to be genetically analysed, the Federal law on human genetic analysis (hereafter LAGH) requires full anonymisation as well as an information and opt-out procedure.
The next question is whether article 321bis and the OALSP apply to (identified or identifiable) biological material. The answer is not evident.
a) On one hand, the reports of the Expert Commission always contain a (short) subchapter on retrospective research on biological material, implying that this research is within its jurisdiction. Moreover, in its guidelines on biobanks, the SAMS has suggested that article 321bis CP and the OALSP extend to biological material, a suggestion that is in line with guidance from the Council of Europe from 2006 [19].
b) On the other hand, the CP and the OALSP explicitly refer to data and this word choice strongly suggests paper or electronic data. To the knowledge of the authors, the Expert Commission has never granted an individual authorisation for research on biological material. Moreover, legal texts tend to mention or regulate data and biologic material separately. In its Message regarding the Federal Law on human genetic analysis, the Federal Council commented on the necessity of adding a provision analogous to article 321bis CP for genetic research. Finally, the policy concerns underlying and justifying data protection measures may not be applicable in the same manner to biologic material.
At present the choice between these two interpretations is not clear-cut. The Swiss federal office for public health still needs to confirm officially whether article 321bis CP and the OALSP also cover biological material used in research. Additionally, it would be essential to know whether reversibly anonymised material (i.e., material which can be re-linked to the patient through a code) should follow the legal regime applicable to fullyanonymised material or that of identifiable material.
What are the other legal options for researchers wishing to access identifiable biological material gathered by a third party? Of course, the ideal way is to ask for patient’s consent. At article 321 CP, the only exception to the rule of consent is an authorisation granted by physicians’ oversight authority. In Geneva, the commission of medical secrecy may be asked to deliver such an authorisation. However, to our knowledge, it has never granted authorisations for research purposes.
It is interesting to refer further to the internal guidelines adopted by the HUG in force since April 2004 (hereafter the HUG 2004 internal guidelines on biological material) [25]. The two fundamental principles of the guidelines are ethics committee approval and patients’ consent. Regardless of whether material is collected from living or dead patients, they (or their representative) must have given prior written consent. Furthermore, the consent must be specific to the research activity contemplated. Exceptions to the principle of consent are only admitted for material collected prior to April 2004 (entry in force of the HUG 2004 internal directive). For such material, the requirements of the HUG 2001 general authorisation are said to apply. This means that a waiver of consent can be requested from the ethics committee if it would be impossible or exceedingly hard to contact patients in order to obtain their consent; additionally, patients must have been informed of the research possibility and not have objected. This second condition is difficult to fulfill given that the general statement on the HUG Web site only refers to data (“données”) and not to biological material. Moreover, the date on which the statement first appeared on the Web site is unknown. It is also unlikely that, prior to April 2004, HUG physicians gave targeted information to their patients about possible research use of their biologic material.
Recently, a working group created by Biobank Suisse, together with the SAMS, suggested a compromise solution for retrospective research on biological samples. It proposes that hospitals obtain systematic general consent from all patients. During a 2009 expert meeting in Bern, the group presented a draft of a model uniform consent form; this document along with accompanying explanations has been circulated for consultation and comments and the final version has been published (http://www.samw.ch/de/Aktuell/News.html).
To make this overview complete, we must envision the situation where physicians go back to their own files and use biological material they collected directly from their patients. The applicable conditions are those mentioned in subchapter A above. In addition, the researchers/physicians must abide by the guidelines established by their research institution. At the HUG, the 2004 guidelines on biological material also apply to retrospective research performed on samples kept by the treating physician (i.e. not accessed from a third party, such as a biobank). This is more strict than what the LPD dictates. The treating physician must obtain written informed consent from the patients (or from their relatives if material is collected from a deceased body). Based on the text of the model form used by the HUG for living donors, this consent applies only to a specific research project. Once consent is obtained, the samples used for the research must be reversibly or irreversibly anonymised.
It is hard to estimate how often research is done by physicians on their “own” biological material. Usually, physicians do not keep this material themselves, but store it centrally in pathology institutes. When this is the case, the samples leave the sphere of control of the physicians. If they want to access them again, we believe that they should follow the rules stated in subchapter C.
After finally agreeing on the wording of a new constitutional article governing research, the Swiss parliament is set to begin discussions on the proposed new law on research (P-LRH) [26]. So far, debates have been heated at every step of the legislative process.
The P-LRH would govern most kinds of health research, including retrospective research on both data and biological material. The text of the new law retains the two cardinal principles underlying all research. Retrospective research would still require the prior approval of an ethics committee and be based on the prior written informed consent of the patient. However, the law would also introduce certain flexibilities and exceptions. The rules and the exceptions contained in the P-LRH are summarized in table 2.
Three points are worth highlighting. First, if the data or the biologic material has been made fully (irreversibly) anonymized (and not just coded), research without explicit consent would be legal, provided that patients were informed of their right to opt out, yet did not exercise it. One problematic aspect of the present P-LRH text is that it uses the term “coded” in a broader sense than the recommendation of the Council of Europe [19]. Under the COE recommendation, if the researcher does not have access to the code allowing to link the biological material to the patient’s identity, the material is said to be “linked anonymized materials”. It then follows the same rules as fully anonymous material or irreversibly anonymized material. The P-LRH does not make a distinction based on who has access to the code. Additionally, it remains to be fully clarified whether material for which the researcher has no access to the code could be given the benefit of the more liberal regime applicable to anonymized material.
Second, retrospective research on (non-genetic) coded data may be performed without (project-specific or project-general) consent, provided the person was informed of this possibility and did not opt-out.
Third, waivers of both consent and prior information about the opt-out right can be granted if the balance of interests favors the research, the patient did not formally oppose such research and it would be impossible or very hard to inform patients or to obtain their consent (or that of their relatives). The waiver conditions are therefore less strict than those actually mandated by article 321bis and the OALSP. In particular, according to the P-LRH, waivers would extend to prior information about veto rights. Perhaps even more importantly, the P-LRH would suppress the authorization mechanism of article 321bis CP and OALSP. The Expert Commission would be disbanded; the general authorizations awarded to hospitals would no longer be needed. The procedure would be replaced by the unique authorization of the ethics committee, which is referred to as “substituted consent”. Hence, ethics committees would decide whether protocols meet the conditions applicable to retrospective research on data or on biological material.
| Table 2Summary of the future rules of the P-LRH on retrospective research. | ||
| – biological material (nonanonymised) | Specific informed consent(valid for the given project) | – impossible or very hard to obtain consent / to inform of veto right; would be painful to the patient to be contacted for purpose of consent or information about veto right+ no document revealing veto of the patient+ the interest of the research prevails over that of the patient |
| – noncoded genetic data | ||
| – coded genetic data | General informed consent (valid for any future project) | |
| – noncoded nongenetic data | ||
| – coded nongenetic data | Right to be informed + veto right | |
| – anonymised data (genetic or not) | P-LRH inapplicable:possible without consent | |
| – anonymised biological material(but not submitted to genetic analysis) | ||
| – anonymised biological material(submitted to genetic analysis) | Right to be informed + veto right already required under LAGH | |
| – small quantity of biological material collected during autopsy or transplantation | No right to be informed + veto right | |
| Step of: | ||
| – anonymisation of nongenetic data | Always freely admitted | |
| – anonymisation of genetic data | Right to be informed + veto right+ no important information could be expected from the research projects | |
| – anonymisation of biological material | ||
Retrospective research should be encouraged. A huge amount of information is collected at university hospitals and it would be regrettable not to use it for the good of research and ultimately patients’ welfare. Electronic medical files represent unique opportunities for large epidemiologic research. Urgent medical questions may be solved by exploiting and linking already available data.
However, because of difficulties in identifying potentially interesting patient data in advance, it is not possible to systematically obtain patients’ advanced informed consent. On this point the mechanism proposed by the OALSP allows for proper balancing of the various stakeholders’ interests. Four suggestions could improve on the present system.
Although international ethics guidelines do not always explicitly require prior information about the possibility to opt out [19, 27], it is crucial, from both an ethical and legal standpoint, that patients be truly informed of their veto right. A simple mention on a gigantic hospital Web site seems hardly sufficient to guarantee this goal. Only a minority of patients are likely to consult the Web site and understand the implications of its short statement regarding research on patient data. The information should at the very least be provided through brochures handed out during treatment and be repeated on posters on hospital walls. Statements on hospital Web sites should be more explicit, visible and multilingual; easy ways to object should be made obvious.
Second, the ethically most crucial point concerning research on biological samples relates to the risk of interfering with the patient’s medical care, should the use of the sample for research purposes jeopardise any potential use for treating or diagnosing the patient. Patients therefore need to be appraised of the risks and must provide informed consent when this hazard cannot be eliminated, in view of the quantities required for research and the nature of the sample (e.g., is it truly a leftover normally thrown away?). The P-LRH should be completed by adding a provision safeguarding this particular interest of patients.
Third, researchers and ethics committees should be more familiar with the conditions under which retrospective research is possible. Some researchers tend to take the oversimplified view that the general authorisation permits any retrospective research, provided the confidentiality form is signed and a study protocol is submitted to the ethics committee [18]. The issue of whether the patient could be contacted is given scant consideration. Too often, waiver is requested simple on the ground that the research is scientifically interesting and patients would be inconvenienced by any contact seeking their consent. This is not however how the law currently reads. Similarly, ethics principles agreed by physicians (e.g., the Helsinki Declaration [28], the SAMS [17] biobank directive) or international bodies [19, 29] do not allow bypassing consent simply because patients would be inconvenienced, would not understand the research or, worse, would refuse to participate. On the contrary, there is near universal agreement that the researcher must show convincingly that it is truly impossible or exceedingly difficult to obtain consent; in some situations, he or she should even begin with a good faith attempt. A few authors have defended the view that requirements for retrospective research on data or biologic material should be relaxed, so as to circumvent consent, trim down information duties and rule out withdrawal of data or material [30, 31]. Their views have encountered opposition from legal and ethical scholars.
Finally, the new P-LRH should be welcomed as it provides a much clearer pathway for retrospective research projects. The level of consent and prior information would be defined in proportion to the risks incurred by patients. Anony-misation (at least reversible anonymisation) would be encouraged. Waivers would be available under simplified conditions. More importantly, the veil of uncertainty surrounding many issues pinpointed in this article would be lifted.
1 WMA: The world medical association declaration on ethical considerations regarding health databases. 2002: http://www.wma.net/en/30publications/10policies/d1/index.html (accessed July 20, 2010).
2 CIOMS: Council for international organizations of medical sciences. International ethical guidelines for biomedical research involving human subjects: http://www.cioms.ch/publications/guidelines/guidelines_nov_2002_blurb.htm (accessed July 20, 2010).
3 Sobel ME: Ethical issues in molecular pathology: Paradigms in flux. Arch Pathol Lab Med. 1999;123:1076–8.
4 Sobel ME, Wolman SR. The ethical uses of human tissues in research. Breast J. 1999;5:153–5.
5 321bis A: Swiss penal code. Available at: http://www.admin.ch/ch/f/rs/311_0/a321bis.html (accessed: July 20, 2010).
6 OALSP: Ordinance regarding authorizations to lift professional secrecy for medical research (hereafter OALSP). Ordinance of the federal council (RS number 235.154) entered into force on July 1, 1993. http://www.admin.ch/ch/f/rs/c235_154.html (accessed July 20, 2010).
7 Expert Commission: Federal expert commission for professional secrecy in medical research. Activity report for years 1998-2000: Http://www.Bag.Admin.Ch/org/02329/02359/index.Html?Lang=fr&download=m3wbpgdb/8ull6du36wenojq1nttjaxznqwfvp7yhmfhnapmmc7zi6rznqckkiv7f3d/bkbxrz6lhudzz8mmps2gpkfo (accessed July 14, 2009). .
8 LPD: Swiss federal law on data protection: Http://www.Admin.Ch/ch/f/rs/c235_1.Html (accessed July 12, 2009).
9 Geneva law on health: Loi sur la santé: Http://www.Geneve.Ch/legislation/rsg/f/rsg_k1_03.Html (accessed July 20, 2010).
10 Expert Commission: Federal expert commission for professional secrecy in medical research. Activity report for years 2005-2007: Http://www.Bag.Admin.Ch/org/02329/02359/index.Html?Lang=fr&download=m3wbpgdb/8ull6du36wenojq1nttjaxznqwfvp7yhmfhnapmmc7zi6rznqckkiz9g3x7bkbxrz6lhudzz8mmps2gpkfo (accessed July 14, 2009).
11 Allen J, McNamara B: Reconsidering the value of consent in biobank research. Bioethics 2009 [Epub ahead of print].
12 Capron AM, Mauron A, Elger BS, Boggio A, Ganguli-Mitra A, Biller-Andorno N. Ethical norms and the international governance of genetic databases and biobanks: Findings from an international study. Kennedy Inst Ethics J. 2009;19:101–24.
13 Elger B. Consent to research involving human biological samples obtained during medical care; in Elger B, Biller-Andorno N, Mauron A, Capron AM (eds): Ethical issues in governing biobanks: Global perspectives. London, Ashgate, 2008, pp. 89–120.
14 Hansson MG, Dillner J, Bartram CR, Carlson JA, Helgesson G. Should donors be allowed to give broad consent to future biobank research? Lancet Oncol. 2006;7:266–9.
15 Hofmann B. Broadening consent – and diluting ethics? J Med Ethics. 2009;35:125–9.
16 ESHG: Data storage and DNA banking for biomedical research: Technical, social and ethical issues. Recommendations of the European society of human genetics. Eur J Hum Genet. 2003;11(Suppl 2):S8–S10.
17 SAMS: Directive on biobanks, obtainment, preservation and utilisation of human biological material. Basel, SAMS, 2006.
18 Data Protection Commissioner: Data protection commissioner’s 2008-2009 report: http://www.edoeb.admin.ch/dokumentation/00445/00509/01551/index.html?lang=de (accessed July 20, 2010).
19 COE: Council of Europe. Rec(2006)4 of the committee of ministers to member states on research on biological materials of human origin: https://wcd.coe.int/ViewDoc.jsp?id=977859 (accessed July 20, 2010).
20 Elger BS, Caplan AL. Consent and anonymization in research involving biobanks: Differing terms and norms present serious barriers to an international framework. EMBO Rep. 2006;7:661–6.
21 Expert Commission: Federal expert commission for professional secrecy in medical research. Activity report for years 2001 to 2004: Http://www.Bag.Admin.Ch/org/02329/02359/index.Html?Lang=fr&download=m3wbpgdb/8ull6du36wenojq1nttjaxznqwfvp7yhmfhnapmmc7zi6rznqckkiv7f3d9bkbxrz6lhudzz8mmps2gpkfo (accessed July 14, 2009).
22 HUG: Hug implementing guideline: Hug intranet. Available: Address request to K. Marescotti.
23 Authorization: Initial general authorization granted to the hug on July 10, 2001, http://www.Admin.Ch/ch/f/ff/2001/3002.Pdf, and its renewed version of 2007, at http://dirmed.Hug-ge.Ch/_library/pdf/autorisationgenerale_2008_2012.pdf.
24 FMH: Conduct code of the Swiss medical association (FMH): http://www.fmh.ch/fr/fmh/bases_legales/code_deontologie.html (accessed July 20, 2010).
25 HUG: Directives du comité de direction sur le prélèvement et/ou l’utilisation d’organes, de tissues, de cellules ou tout autre matériel biologique sur l’être humain à des fins de recherche ou d’enseignement. Genève, Hôpitaux Universitaires de Genève, 2004.
26 Swiss federal government: Swiss federal government: Steps of the law project: Http://www.Parlament.Ch/f/suche/pages/geschaefte.Aspx?Gesch_id=20070072 (accessed July 20, 2010).
27 WMA: World medical association. Declaration of Helsinki, adopted by the 59th wma general assembly, Seoul, October 2008: Http://www.Wma.Net/e/policy/b3.Htm (accessed July 14, 2009).
28 WMA: World medical association. Declaration of Helsinki, adopted by the 52nd world medical association general assembly, Edinburgh, Scotland.
29 WHO: Who’s guideline for obtaining informed consent for the procurement and use of human tissues in preparing a research project proposal third edition – 2000: Http://www.Who.Int/reproductivehealth/topics/ethics/human_tissue_use.Pdf or (entire document): Http://www.Who.Int/reproductivehealth/topics/ethics/human_tissue_use_guide_serg/en/index.Html (accessed July 14, 2009).
30 Hansson MG. Combining efficiency and concerns about integrity when using human biobanks. Stud Hist Philos Biol Biomed Sci. 2006;37:520–32.
31 Eriksson S, Helgesson G. Potential harms, anonymization, and the right to withdraw consent to biobank research. Eur J Hum Genet. 2005;13:1071–6.
Both authors are members of research ethics committees at the Geneva University Hospitals (HUG) and therefore review study protocols, including retrospective studies.
There are no conflicts of interest.
Funding was provided by a research grant for advanced researchers of the National Science Foundation for B. Elger